")
")
| Issue |
Sci. Tech. Energ. Transition
Volume 78, 2023
|
|
|---|---|---|
| Article Number | 32 | |
| Number of page(s) | 60 | |
| DOI | https://doi.org/10.2516/stet/2023029 | |
| Published online | 28 November 2023 | |
Review Article
Literature review: state-of-the-art hydrogen storage technologies and Liquid Organic Hydrogen Carrier (LOHC) development
1
Univ. Grenoble Alpes, 38000 Grenoble, France
2
CEA, LITEN, DTNM, 38054 Grenoble, France
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
10
March
2023
Accepted:
4
September
2023
Abstract
Greenhouse gas anthropogenic emissions have triggered global warming with increasingly alarming consequences, motivating the development of carbon-free energy systems. Hydrogen is proposed as an environmentally benign energy vector to implement this strategy, but safe and efficient large-scale hydrogen storage technologies are still lacking to develop a competitive Hydrogen economy. LOHC (Liquid Organic Hydrogen Carrier) improves the storage and handling of hydrogen by covalently binding it to a liquid organic framework through catalytic exothermic hydrogenation and endothermic dehydrogenation reactions. LOHCs are oil-like materials that are compatible with the current oil and gas infrastructures. Nevertheless, their high dehydrogenation enthalpy, platinoid-based catalysts, and thermal stability are bottlenecks to the emergence of this technology. In this review, hydrogen storage technologies and in particular LOHC are presented. Moreover, potential reactivities to design innovative LOHC are discussed.
Key words: Hydrogen / Hydrogen storage / Liquid Organic Hydrogen Carrier (LOHC) / Hydrogenation / Dehydrogenation / Heterogeneous catalysis
© The Author(s), published by EDP Sciences, 2023
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
Access and use of external energy have always been a preoccupation of human societies to facilitate the satisfaction of basic needs such as food cultivation/preparation, warmth production, or crafting of everyday materials. Our ancestors had simple energy forms at their disposal such as human muscle, animal muscle, burning of biomass, and renewable energy derived from the wind, water, and sun [1]. While used for tens of millennia, the development of stationary steam-powered engines supplied by fossil fuels in the late 18th century revolutionized the means of production. The development of new and more efficient processes like the replacement of wood for coal in the iron industry, deeper mining, or chemicals (especially sulphuric acid and sodium carbonate) durably modified Western societies, leading to the Industrial Revolution [2–6]. Since then, a strong increase in the global atmospheric concentrations of greenhouse gases such as carbon dioxide (CO2), methane (CH4), and nitrous oxide (N2O) was observed and their heat-trapping property started to abnormally modify the global climate, being later dubbed as climate change [7]. Over the past decades, this increase has been linked to anthropogenic emissions related to the combustion of fossil fuels (coal, oil, gas) for CO2 and agricultural malpractices for CH4 (manure and biomass burning) and N2O (synthetic inorganic fertilizers) [8–10]. As the effects of climate change are already visible and partially irreversible, the rapid reduction of greenhouse gases is a key target to limit the dire consequences on ecosystems, biodiversity, and human societies [11]. Under these circumstances, new carbon-free energy systems must be developed in order to simultaneously tackle the reduction of greenhouse gases and the access to energy for the already energy-lacking hundreds of millions in Africa, South America, and South–East Asia, while anticipating a global demographics increase [12].
Electricity is an energy sector where low-carbon alternatives such as nuclear power and renewable energies (namely solar power, wind power, hydropower, and geothermal power) are already implemented [13]. Low-carbon energies have gained traction over the last decades, earning from their much-added benefits to safety and direct CO2 emission reductions. The CO2 emission factor obtained by Life-Cycle Analysis for each power generation source without carbon capture is presented in Figure 1 [14, 15].
 |
Figure 1 Direct and indirect CO2 emission factors for different types of power generation systems obtained by life-cycle analysis and adapted from [14, 15]. |
While the production of electricity can be attained by low-carbon technologies, it is important to note that 84% of the global primary energy production still came from fossil fuels in 2019 [1]. Hence a strong electrification of our societies is proposed to further the use of low-carbon technologies. As a consequence, the part of renewable energy sources has been steadily increasing from 14% to 22% in the French energy mix between 1990 and 2021, aiming for 32% in 2030 (Fig. 2) [16–18].
Although this energy transition has mainly consisted of reducing the part of nuclear power in the energy mix, new challenges are arising from this situation. Indeed, most of the prevalent renewable solutions (sun power, wind power, hydropower) are intermittent, i.e. they depend on the weather or a time cycle (day/night, seasonal, etc.) (Fig. 3) [19].
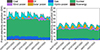 |
Figure 3 Energy production in France in January 2022 (left) and July 2022 (right), adapted from [20]. |
The energy variations due to the renewables are currently compensated by the pilotable energies (fossil fuels and nuclear). However, with their planned decrease in the energy mix, it will be progressively more and more difficult to adjust the production/consumption ratio and, in a hypothetical 100% renewables scenario, such a compensation mechanism would be strictly impossible. Moreover, excess energy produced by renewable sources during fast transitional regimes is currently discarded, leading to a net energy loss [20–22]. Hence, developing a technology to store the excess renewable energy for later use is key to removing pilotable fossil fuels from the energy mix and successfully abating greenhouse gas emissions. In addition, energy uncertainty management methods demand the dynamic integration of energy storage systems in the electric power distribution networks and the probable oversizing of the renewable power capacity [23, 24].
Thus, the design of smarter energy systems is sorely needed. An energy system is characterized by a succession of operations such as storage, transportation, conversion, and transformation of a set of energy resources to produce fine usable energy for a desired purpose (e.g. heat up our house). Following this example of our everyday life in a cradle-to-gate approach, an energy resource (for example coal) is extracted from the ground, then refined (conversion), stored, transported to a coal power plant, transformed into electricity which is then transported to our house before being converted to heat by a resistance in a heater (Fig. 4). It is worth noting that each step can produce waste and interact with the environment so limiting the number of steps usually is an efficient method to minimize adverse effects.
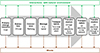 |
Figure 4 Example of an energy system to produce heat through a heater from coal. |
The transport and storage of energy are two essential steps for energy systems as energy is often produced remotely. Moreover, the need for instantaneous and continuous energy output requires additional energy to be stored in order to back up the energy systems in case of supply disruption. In our previous example, coal is a solid that can easily be stored and transported before use. However, swapping coal for renewable energy like solar power drastically changes our energy system as storing vast amounts of electricity becomes much more complex than a solid (Fig. 5).
 |
Figure 5 Example of an energy system to produce heat from a heater from solar radiation. |
The concept of the energy vector answers this issue: the energy is converted into a new form that can be easily stored and transported. It is crucial to make a clear distinction between the primary energy source and the energy vector. In the energy system chain, a primary energy source such as fossil fuels, nuclear power, and renewable energies directly produces energy: this energy is readily available and will be lost if not used or stored otherwise. Primary energy resources can be separated into either renewable energy resources (solar radiation, water, biomass, wind, earth, sea, biological, and bacterial) that can be re-formed in the environment by natural processes or non-renewable resources (coal, oil, gas, and nuclear fuels) that cannot be regenerated by the environment in a period of time comparable to their use. Alternatively, a potential definition of an energy vector is human-made energy that is intended to replace a primary energy source at a required place and time [25]. In recent history, coal, oil and natural gas have been the go-to energy vectors backing up most of our energy systems. Other energy vectors include electricity, hydrogen (H2), synthetic fuels (methanol, ethanol, biodiesel, biogas, syngas), heat transfer fluids (low viscosity mineral oils), mechanical vectors (mechanical, oil-dynamic, and pressure-dynamic transmissions) and radiation. Each energy vector transports energy in a different form, either chemical, electric, thermal, mechanical, or radiative. In addition, energy vectors are not strictly replaceable with one another as each possesses its intrinsic limitations on its transportation and storage potential. A brief overview that does not take into account the storage efficiencies of the mentioned energy vectors’ transportation and storage capabilities is found in Figure 6.
 |
Figure 6 Transport range and storage time of energy vectors. Electricity and radiation cannot be directly stored and thus are not included in the diagram. |
Among the energy vectors, H2 possesses a transport range and a storage time comparable to the currently employed fossil fuels depending on the employed storage technology. Mechanical systems based on physical phenomena such as compression used in Compressed Air Energy Storage (CAES) [26] or potential energy for Pumped Hydro Storage (PHS) [27] and heat exchange fluids can efficiently store energy, but their transportation properties of the stored energy are limited. Conversely, while electricity can be easily transported by the current power distribution network in the form of electrons, it can only be stored in limited amounts by supercapacitors with high self-discharging rates [28]. Hence conversion of electricity is usually needed to indirectly store it in electrochemical devices such as batteries (Li-ion, Pb acid, NaS, etc.) [29]. Finally, radiation such as light cannot be stored; it has however the theoretical highest transport range in vacuum, making it the best energy vector for interplanetary energy transfer [30].
2 Hydrogen as an energy vector
As presented in Figure 6, H2 presents transportation and storage properties similar to fossil fuels such as natural gas. Another interesting characteristic of H2 is its simple reactivity with O2 to yield only water and heat as products of the reaction, making it virtually an environment-friendly technology (1).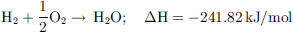 (1)
(1)
Moreover, as seen previously, the implementation of intermittent renewable energies imposes the storage of large quantities of energy (in the TWh range) over a long duration (monthly to yearly) to cover seasonal variations. Fortunately, H2 possesses a high mass-energy density of 120 MJ/kg, about three times more than methane and other hydrocarbons [31, 32]. However, as H2 is a gas at normal temperature and pressure, it presents greater barriers to implementation compared to current liquid fuels: its density is minimal (0.09 g/L under normal conditions of temperature and pressure), leading to a necessary concentration of H2 in order to limit the size needed for storage [33]. Other characteristic disadvantages of H2 are its small size which facilitates its permeation out of containers, its embrittlement-inducing reactivity with common pipeline materials as steel, and potential ozone depletion if released in significant quantities [34–37].
Despite these limitations, H2 is globally considered an energy vector able to store vast amounts of energy over a long time scale that can be used as a secondary energy source for out-of-network applications and intercontinental storage and transportation energy systems. Promoted by newly implemented carbon regulations, H2 is becoming an opportunity to achieve a clean and secure energy future and its deployment is supported by different policies in various countries (Fig. 7) [38].
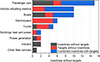 |
Figure 7 Number of policies around the world supporting hydrogen deployment, International Energy Agency, 2018 [38]. |
From this assessment, it is clear that building a hydrogen economy faces mainly financial and technological barriers. In order to boost its competitiveness, it is primordial to assess the key steps of the H2 economy value, namely its production and storage. In this review, the currently available H2 production technologies will be briefly covered before focusing on the available and in-development H2 storage technologies.
3 H2 generation
Except in some rare occurrences, pure H2 is unavailable in nature as it can break from Earth’s gravitational pull due to its low density and then leave the atmosphere when escaping from the underground [39–41]. Therefore, H2 needs to be produced from resources available in the environment. Current H2 production principally relies on converting non-renewable feedstock such as coal, Natural Gas (NG), and oil while only 5% is produced by renewable sources like water electrolysis and biomass [42, 43].
This section will review the different technologies available to produce H2 and their potential with regard to the energy transition.
3.1 Non-renewable H2 production (grey/brown hydrogen)
On the non-renewable side, three main reforming processes are currently employed to produce H2 and Carbon Monoxide (CO) from fossil carbonaceous substrates in the presence of a catalyst: Steam (Methane) Reforming (SMR), partial oxidation of hydrocarbons and autothermal reforming [44–47]. Further refinement of CO can be performed by the Water-Gas-Shift Reaction (WGSR) in order to produce a supplementary equivalent of H2 and CO2. Here, the main issues with these processes come from the concomitant production of CO2 as well as their high energetic cost due to the high pressure (up to 100 bar) and temperature (500–1500 °C) required for the reactions to occur. Although Carbon Capture and Sequestration (CCS) at the exhaust is getting more prominent, it does not intrinsically solve the thermodynamics issue and the efficient valorization of the CO2 is yet to be addressed [48]. More recently, hydrocarbon pyrolysis is being rapidly developed as it is in principle able to cleanly produce solid carbon and H2 at a reduced energy cost (38 kJ/molH2) compared to water electrolysis (285 kJ/molH2) (see Sect. 3.2) [49, 50]. As no CO2 is formed during the process and solid carbon can be valorized in different fields (e.g. soil enhancer), this technology is promising and companies like BASF are planning for industrialization in 2025 [51]. However, critics are also rising due to the use of methane (CH4) as hydrocarbon feedstock. Indeed, as CH4 emissions play a huge role in global warming, hydrocarbon pyrolysis would yield beneficial effects only if the global CH4 emissions diminish faster than the decarbonization of electricity [52]. Finally, all hydrocarbon-based processes are dependent on the quality of the feedstock. In particular, sulphur can poison the catalytic surfaces and react with H2 to form H2S, decreasing the purity of the gas and the overall efficiency of the reaction.
3.2 Water electrolysis (blue hydrogen)
Water electrolysis was discovered in 1789 by Jan Rudolph Deiman and Adriaan Paets van Troostwijk by producing electrostatic discharge between electrodes immersed in water [53]. While industrial interest sparked in the 1920s and 1930s, only recently has it been rediscovered as a convenient way to convert the intermittent electricity produced by renewable energies [54]. Water electrolysis is considered eco-friendly due to the lack of direct CO2 emissions in the process. However, the electric sources used to power the cell and the significant amount of required power play a huge role in the environmental impact of the process (hydro, wind, sun, nuclear versus gas, oil, coal) [42, 55]. As the price of renewable electricity is plummeting, decarbonization plans in Europe are promoting water electrolysis as the leading technology to produce renewable H2. In the wake of this trend, large-scale electrolysis facilities are being built around the world. The worldwide electrolyser capacity was 11 GW in 2022 with a possible installed capacity of 170–365 GW in 2030 if all of the planned projects are carried out. However, as an electrolysis capacity of 550 GW was envisioned by 2030 in the Net Zero Emissions scenario, a drastic increase of the electrolysis capacity installation is needed to achieve this target [56].
Water electrolysis is an umbrella term for different electrolysis technologies, namely alkaline [57], polymer electrolyte membrane technologies based on the transfer of protons (PEM) [58], or anions (AEM) [59] and Solid Oxide Electrolyte (SOE) [60]. On a general basis, these technologies are based on the application of an electric current to dissociate the atoms of H2O and then recombine them to form high-purity H2 and O2 (2).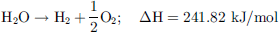 (2)
(2)
From a thermodynamic standpoint, this reaction is endothermic and nonspontaneous (see (1)). In standard conditions, a thermodynamic potential of 1.23 V is sufficient to produce H2. However, the irreversible energy costs associated with the operation of the electrolysis cell must be taken into account like the slow formation of O2 compared to H2 and ohmic losses due to resistances in each component of the circuit, in particular the resistance of the electrolyte. Therefore, the power-to-H2 efficiency depends on the employed technology: 60–70% for alkaline electrolysis, 60–80% for PEM electrolysis, and 40–60% for SOE electrolysis [61]. Nonetheless, with a supplied external heat of 150–180 °C to generate steam, up to 95% power-to-hydrogen efficiencies could be obtained for SOE electrolysis [62, 63]. Finally, alkaline and PEM electrolysis are currently the only commercially available technologies while SOE electrolysis is yet to be fully developed.
3.3 Renewable H2 generation (green H2)
Renewable sources of H2 can be separated into distinct classes as H2 can be obtained by transformation of a renewable resource by thermochemical or biological processes. It is worth noting that most renewable H2 sources can act as energy storage systems but of low capacity and/or only on a short-term basis.
Biomass usage possesses numerous beneficial environmental, social, and economic aspects such as the restoration of degraded lands, poverty reduction, and CO2 entrapment in carbon wells. Its conversion through various processes holds great promise for sustainable H2. Biological processes are based on anaerobic digestion [64] and fermentation followed by alcohol reforming [65, 66]. These technologies are promising but work only in optimal conditions and/or with little output. Other renewable H2 technologies revolve around biomass thermochemical processes such as pyrolysis [67], gasification [68], and supercritical water gasification [69]. As thermochemical processes are closer to the current industrial processes, they gather great interest. However, as numerous intermediate species can be produced, liquid and gas purification costs might be a major hurdle for these technologies.
3.4 Cost comparison of H2
The cost comparison of the different technologies used to produce H2 is presented in Figure 9 [70–79].

As numerous apparatus exist for each process, some variations in the hydrogen cost can be found for the same technology. Currently, the combined technologies SMR + WGSR without CCS are the cheapest energy source to industrially produce H2. However, H2 is nowadays proposed to store the excess of intermittent renewable energies. Currently, most of the produced H2 originates from the SMR + WGSR process and H2 produced from renewable is not competitive. Here, a decrease in the price of H2 generated from renewable energy is expected due to the massive implementation of renewables. Moreover, the SMR + WGSR process should be disfavored over time due to the emergence of new environmental legislation and the upsurge of global tensions.
Nevertheless, most techno-economic analysis in the literature limits their framework to the economic aspects of the technologies. To overcome this bias, a recent contribution from Al-Qahtani et al. suggests taking into account external factors in the price of H2 such as their costs on human health, ecosystem quality, and resource depletion by using life cycle monetization. The cheapest energy source for H2 production was still SMR + WGSR with direct carbon capture for 5 $/kgH2. Nevertheless, the most efficient energy sources with regard to the external factors were nuclear power, wind power, and solar power where 86%, 77%, and 86% respectively of the H2 cost was due to the selected technology [80]. The cost reduction of renewables and incentives should then play a key role in increasing their competitiveness for H2 production.
4 State-of-the-art hydrogen storage technologies
Once produced, H2 needs to be stored if not readily used. However, economic H2 storage is not straightforward due to its very low density: 11 m3 (size of the trunk of a big utility vehicle) would be required to store 1 kg of H2, whose energy is equivalent to driving a car for 100 km. Many H2 storage technologies aim at concentrating H2, based either on the physical properties of H2 (compressed gas (CGH2), cryogenic liquid (LH2) and Underground Storage Systems (UHS)) or on chemical properties of materials (physical adsorbents, metal hydrides, B–N H2 carriers, circular H2 carriers, and liquid organic H2 carriers) as presented in Figure 10.
 |
Figure 10 Reviewed H2 storage technologies. |
Each technology will be briefly reviewed before focusing on Liquid Organic Hydrogen Carriers (LOHC) as well as other acceptorless hydrogenation and dehydrogenation reactions. Technologies can always be compared by the H2 weight storage density (wt.%H2) and the H2 volumetric storage density (gH2/L), but, depending on the encompassed element of the storage system, the said densities can be drastically modified. Here, we will only focus on the theoretical maxima of the hydrogenated species for chemical systems.
4.1 Physical-based hydrogen storage
Physical H2 storage technologies are based on controlling external parameters such as the pressure and temperature to concentrate H2, leaving the H2 molecule unmodified.
4.1.1 Compressed gas H2
Compressed gas H2 in steel cylinders is the most frequent H2 storage technology, storing 200 bar H2 (16.8 gH2/L, without the weight of the system). In theory, a continuous increase of the H2 pressure is the simplest way to improve the storage efficiency, providing that pressure-resistant materials are available. However, the H2 volumetric density is not linearly correlated with its pressure, and technical as well as economic restrictions limit the compression well below pressures equivalent to cryogenic H2 storage (2000 bar, 70 gH2/L, see 4.1.2) [81]. As a compromise between energy need and costs, the industry targets a pressure of 700 bar H2 with a total H2 and container system mass of 125 kg (including H2) using lightweight polymer fiber-reinforced vessels. Such systems could reach a weight density of 5 wt.%H2 and a volumetric density of 30 gH2/L, appropriate for personal vehicle transportation [82]. While this solution is practical and aimed at various fields such as heavy-duty transportation, it poses safety issues in case of violent breach of containment or uncontrolled H2 accumulation in an enclosed space like underground parking [83]. Finally, reaching a pressure of 700 bar of H2 has a non-negligible energy cost. In ideal isothermal conditions, 2.21 kWh/kgH2 is needed to compress H2 from atmospheric pressure to 700 bar. As the compression is not isothermal in real conditions due to self-heating upon applying pressure, the compression cost is significantly higher (up to 4 kWh/kgH2, more than 10% of the energy stored) [84, 85]. CGH2 presents numerous risks and limitations, but the relative simplicity of this technology explains its high prevalence for mobility applications.
4.1.2 Liquid H2 (LH2)
Hydrogen is a gas that reaches its liquefaction point at −252 °C, with a volumetric density of 70.8 gH2/L and a system weight density of up to 90 wt.%H2 depending on the container [86]. However, the liquefaction of H2 is not trivial due to its specific properties (inverse Joule-Thomson effect) [87]. Moreover, gas phase H2 is present in two forms depending on the nuclear spin alignment of its individual atoms (75% ortho) or not (25% para). Conversely, liquid phase H2 is stable at 99.8% in the para form, leading to the necessary conversion from ortho to para [87]. Unfortunately, this conversion is slow and exothermal (∆H = 525 kJ/kg), inducing an unavoidable evaporation of H2 (latent heat of vaporization = 476 kJ/kg), also called boil-off [88]. To facilitate this slow conversion during the cooling process, a catalyst is often integrated into the heat exchanger [89]. If this phenomenon is not accounted for, the pressure could drastically increase in a closed system, hence LH2 storage must be kept open to avoid disastrous events [90]. Open systems raise the question of efficient thermal insulation, with current systems losing roughly 1.5 wt.%H2/day. Finally, current industrial liquefaction plants consume between 13 and 15 kWh to produce 1 kg of LH2 (40–45% of the stored energy) and are not expected to drop below 5 kWh/kg LH2 (15% of the stored energy) [87]. Thus, the energetic cost of liquefaction is prohibitive for large-scale deployment and, while early projects targeted car mobility, LH2 will probably be used only for specific energy-intensive applications like rockets [91–93].
4.1.3 Underground H2 storage
Underground H2 storage is a potential large-scale mid to long-term stationary storage technology. Underground geological structures like empty gas or oil reservoirs, aquifers, and salt caverns can act as gas vessels to store pressurized pure H2 or a mixture of gases that contain H2. An appropriate UHS possesses a solid rock formation and an inert impermeable layer to prevent H2 leaks as H2 can slowly react with the minerals composing the UHS to form carbonates [94]. UHS also have to withstand an internal pressure of 30 to 80% of the lithostatic pressure in order to limit H2 leaks [95]. For the time being, only four artificial salt caverns are exploited worldwide [96]. For example, the most massive salt cavern (Spindletop, USA) occupies a volume of 906 000 m3 for a pressure reaching up to 200 bar (16.8 gH2/L) and could in theory store up to 500 GWh of H2, roughly 0.02% of the yearly French primary energy consumption in 2020 [97]. Salt caverns are the most efficient UHS, but represents only 8% of the worldwide capacity. Finally, while highly promising, UHS is especially sensitive to geological events like earthquakes, challenging to conceive/monitor, limited to specific geological conditions, and is in competition with other underground gas storage systems like CAES or CO2 storage.
4.2 Chemical-based hydrogen storage
The modification of the physical properties of H2 such as its temperature or pressure increases its energy density but requires a high energetic cost in order to reach efficient storage. A decrease of this energetic cost (equivalent to more than 10% of the energy stored for 700 bar CGH2 and between 40 to 45% for LH2) is necessary if H2 is to become a competitive energy vector. Hence finding new materials able to store H2 by forming bonds ranging from weak van der Waals interactions to covalent bonding has been an ongoing topic of research over the past decades. The main technologies of chemical-based storage will be reviewed hereinafter.
4.2.1 Physical sorbents
Physical sorbents can be compared to sponges for gaseous H2 where molecular H2 is adsorbed by physisorption on the surface of microporous materials by van der Waals interactions. H2 adsorption is usually carried out at a cryogenic temperature and high pressure (20–200 bar) for better performance. However, these materials must be kept at cryogenic temperatures and high pressures in order to be able to store high quantities of H2, increasing over time the energy cost of the storage. Moreover, the H2 intake is not linear with the increased pressure as strong adsorption is usually only observed at low pressures (up to a few bars) where capillarity or monolayer phenomena take place in the structure micropores. The intake increases slowly at higher pressures as all micropores are filled or multiple gas layers start to stack on top of each other, creating a non-adsorbed gas phase [98]. By heating up the material to room temperature or above, pure H2 can easily be released with only small amounts of H2 trapped in the structure. Finally, serious doubt has been cast on the validity of early results of physical sorbents for H2 storage. Indeed, the lack of reproducibility and high gravimetric H2 storage were linked to gas leaks and to the adsorption of molecules of higher molecular weight like water [99]. Current materials used for the physisorption of H2 are carbon nanomaterials (nanotubes, activated carbon, nanofibers, fullerenes, and so on), Polymers of Intrinsic Microporosity (PIM), Covalent-Organic Frameworks (COF), Metal-Organic Frameworks (MOF), zeolites, and water clathrates. The characteristics of each family of materials will be briefly described.
4.2.1.1 Carbon nanomaterials
Carbon Nanotubes (CNT), activated carbon, nanofibers, fullerenes, and graphenes are able to store H2, but this property is heavily dependent on their structure, geometry, operating temperature, pressure, and other parameters of the material. These materials possess multiple complex H2 adsorption sites that lead to extremely variable H2 capacity with a theoretic maximum of 10.8 wt.%H2 at 77 K and 60 bar [100]. However, it has been shown that the same material can produce inconsistent results. For example, a K-doped CNT was reported to store 1.8 wt.%H2 and 14 wt.%H2 in the same operating conditions [101, 102]. In addition, work by General Motors reveals that all H2 capacities above 1 wt.%H2 are probably due to experimental errors attributed to leaks [103]. Thus, the usual storage capacity at room temperature and high pressure is usually below 2 wt.%H2 [104]. Higher storage capacities can be reached when adsorption is conducted at cryogenic temperatures, with capacities of 6.5 wt.%H2 reported for activated carbon [105]. Chemisorption as π-bonding on aromatic cycles is also possible with extreme pressures (>500 bar) and high temperatures (500–600 K) treatments, adding one more H2 molecule per aromatic cycle [106]. Desorption happens around 400 K, but the extreme conditions for H2 chemisorption prevent it from being a viable H2 storage method [107].
4.2.1.2 Polymers of intrinsic microporosity
Microporous Polymers (PIM) are able to store H2 due to their rigid structure that forms interconnected cavities in the nanometer range, facilitating H2 adsorption [108]. Their intrinsic advantages are their easy customization by varying the monomer structure, their lightness, low cost, and simplicity of process. Nonetheless, they suffer from disadvantages similar to those of carbon nanomaterials as they require cryogenic temperatures and higher pressures to store relevant H2 amounts. Indeed, PIM can absorb up to 1.4 wt.%H2 at 1 bar and 77 K, increased to 1.7 wt.%H2 at 10 bar [109]. PIM showed improved properties when mixed with another physical sorbent, exhibiting up to 2.5 times more H2 adsorption, and is the current research focus for this technology [110].
4.2.1.3 Covalent-organic frameworks and metal-organic frameworks
Covalent-Organic Frameworks (COF) are rigid porous structures composed of organic molecules that are linked together by covalent bonds and are used for gas adsorption, separation, and catalysis like pesticide degradation [111, 112]. Due to their organic nature, COFs are extremely light, low cost, highly stable, and show great structure versatility in both 2D and 3D networks with different cavity sizes able to accept various molecules [113]. H2 adsorption in the structure can be achieved, usually at cryogenic temperatures (77 K) and at high pressures (20–100 bar). Temperature is the main factor contributing to their performance, as increasing the temperature above cryogenic levels drastically reduces their H2 adsorption capacity. To this date, a variation of the COF-102 showed very high gravimetric and volumetric densities of 25 wt.%H2 and 43 gH2/L at 77 K and 100 bar. However, room temperature H2 adsorption measurements on the same material exhibited reduced densities of 6 wt.%H2 and 10 gH2/L [114]. Doping the structure with metal ions was proposed in computational studies to improve the H2 capacity at room temperature, but no practical study has been reported so far [115].
Similar to COF, Metal-Organic Frameworks (MOF) are composed of metallic single atoms or clusters linked together by organic molecules, forming a porous crystalline structure able to adsorb H2 at cryogenic temperatures (77 K) and at high pressures (20–100 bar) depending on the cavity sizes and affinity with the metal and/or the organic linkers [116]. The current best H2 adsorption MOF is DUT-32 with a gravimetric density of 14.21 wt.%H2 at 77 K and 82 bar [117]. Addition of a metal catalyst in the structure and porous support like activated carbon could favor hydrogen spillover, where H2 is split into atoms and incorporated into the support, allowing for room-temperature H2 storage. While numerous publications reported a significant improvement of the hydrogen storage properties, spillover was heavily dependent on a number of inconsistent parameters, creating irreproducibility and even questions about the improvement of MOF by spillover [118–120].
4.2.1.4 Zeolites
Zeolites are crystalline materials defined by their network of pores that have been used for decades as sorbent materials and molecular sieves [121]. H2 adsorbing zeolites are composed of aluminosilicate porous structures with dimensions comparable to that of carbon nanotubes. Moreover, the charge of the framework is counterbalanced by metal cations that are readily exchangeable, which enables the tuning of the properties of the zeolite. H2 adsorption is believed to follow two mechanisms depending on the temperature. At high temperatures, encapsulation is the preferred mechanism: the openings of the zeolites are thermally activated which forces H2 adsorption in the structure, and the following cooling traps it [122, 123]. However, this mechanism does not substantially increase the amount of H2 stored in the structure as only a maximum of 0.8 wt.%H2 adsorption was reported [122]. At room temperature, the zeolite entrances were not activated, requiring high pressures to force H2 in the structure, leading to poor performances (<0.1 wt.%H2) [123, 124]. Better results were obtained when cooling the zeolite to cryogenic temperatures, with a maximum of 2.19 wt.%H2 for a CaX zeolite at 77 K and 15 bar [125]. Nevertheless, the theoretical maximum storage capacity was calculated to be less than 3 wt.%H2 which limits the efficiency of this storage technology [126].
4.2.1.5 Clathrates
Clathrates are supramolecular materials able to trap H2 in cavities formed by water molecules, without the formation of covalent bonds [127]. Pure H2-water clathrates can be formed and stabilized at cryogenic temperature (77 K) or extreme pressures (150 bar to 23 kbar reaching densities of 5.5 wt.%H2 and 45 gH2/L [128, 129]. The addition of a small amount of promoters like tetrahydrofuran (THF) stabilized the clathrates while limiting the temperature and pressure conditions for storage at the cost of an H2 storage capacity reduced to 1–2 wt.%H2 or less depending on the pressure [130]. Except for pure H2-water clathrates that are formed and stored in extreme conditions, the performance of hydrates clathrates does not deviate from other physical sorbents.
4.2.2 Metal hydrides/Interstitial hydrides
Metal hydrides are solid-state materials that attracted very early attention as a mean of hydrogen storage for individual transportation due to the reversible H2 absorption properties of metals M at high temperatures in less than an hour following the equation (3) [131]. The dehydrogenation reaction produces at the same time scale the original metallic phase and H2 with its kinetics being controlled by the temperature, pressure, and presence of a catalyst [132]. In addition, some Mg-based metal composite hydrides can be cycled up to 2000 cycles with good reversibility without major decrepitation due to the scaffolding effect of the composite [133].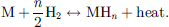 (3)
(3)
In general, hydrides are heavy materials with low gravimetric and volumetric densities. Moreover, they are air- and water-sensitive (pyrophoricity) as oxygen tends to remove surface hydrogen to produce oxides, hydroxides, and carbon-oxygen compounds in the form of a surface passivation layer. Removal of this layer with H2 at high temperatures is necessary in order to increase the hydrogenation kinetics [134–136]. Sulphur compounds are also a common poison [137]. Finally, these materials consume a non-negligible amount of metals and their solid state complicates their handling from an industrial standpoint as their processing consumes more energy than liquids and can form reactive and hazardous dust.
Metal hydrides can be grouped into low-temperature hydrides, high-temperature hydrides, and complex hydrides depending on their desorption temperature and composition. A brief overview of the properties of each class of materials will be presented.
4.2.2.1 Low-temperature hydrides
Low-temperature metal hydrides, also known as intermetallic hydrides, can release H2 close to room temperature and atmospheric pressure, which is advantageous as almost no energy is required to harness H2 but also a drawback due to safety issues in case of undesired heating [138]. Intermetallic hydrides are synthesized from a mixture of high hydrogen affinity elements “A” like Ca, Sc, Y, Ti, Zr and other lanthanides and low-affinity elements “B” like Cr, Mn, Fe, Co or Ni that act as dissociation promoters to create ternary A x B y H n materials [139]. During the absorption, H2 is dissociated in H atoms on the surface and form covalent bonds before migrating in the bulk of the material by atomic diffusion to the interstitial sites of the lattice [134]. Therefore, the defined crystalline structures (AB5, AB2, A2B, …) play a primordial role in the H2 adsorbing properties as the size of their interstitial sites is phase-dependent [140]. However, due to their weight, the maximum energy density of intermetallic hydrides stays usually below 2 wt.%H2, far from the necessary 6 wt.%H2 for mobility applications according to the United States Department of Energy (DOE) [141, 142].
4.2.2.2 High-temperature hydrides
High-temperature metal hydrides require desorption temperatures above 200 °C to break the ionically bound hydrogen atoms. Most high-temperature hydrides are based on magnesium and its alloys due to its lightness, abundance, low cost, and good reversibility. The most studied one is MgH2, which possesses a high weight density of 7 wt.%H2. Unfortunately, this material requires a temperature of at least 300 °C to harness H2 due to its high stability, slow kinetics, and sensitivity to decrepitation [143, 144]. Alloys of high-temperature metal hydrides with transition metals like Ti, V, Mn, Ni, and Fe have shown improved thermodynamics, H2 uptake/release kinetics and stability by promoting the dissociation/recombination of H2 [145]. Nanostructuring the metal hydride is also a conventional technique to decrease the size of the metal clusters to the nanometer range in order to improve the thermal and mechanical stability, thermodynamics and kinetics [146–148]. Finally, akin to nanostructuring, nanoconfinement relies on the nano-scale properties of hollow light-weight nano-porous chemically inactive materials like carbon-based materials, mesoporous silica, zeolites, and MOFs to promote H2 physisorption, H2 dissociation, desorption thermodynamics, and kinetics as well as stability by limiting the agglomeration of the metal hydride nanoparticles [149–151].
4.2.2.3 Complex hydrides
Over the past two decades, complex hydrides containing only low molecular weight atoms have been heavily studied [152]. Compared to the desorption temperature of low-temperature metal hydrides (room temperature to 100 °C) and high-temperature metal hydrides (200–300 °C), their intermediate desorption temperature (100–200 °C) presents a good compromise between safety and energy efficiency. Such desorption temperatures are achieved by exploiting the versatile nature of the hydrogen atom that can act as both a cation H+ and an anion H−. Indeed, H2 is stored in complex hydrides by a mix of ionic and covalent bonds, usually forming tetrahedrons with boron or aluminum at the center and hydrogen atoms at the corners while the charge is balanced by one or more spectator cations like Li or Na. The dehydrogenation process is heavily dependent on the metallic center, however, it typically consists of the formation of a pure metallic phase and a cation-hydride phase, e.g. in the case of alanates (4):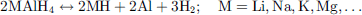 (4)
(4)
In general, complex hydrides are synthesized by mechano-chemistry like ball milling which limits their industrialization. In addition, they present a high chemical risk as they are highly reactive in the presence of water and must be used only in anhydrous conditions [153]. Moreover, reversibility can be limited due to the lack of hydrogen-deficient intermetallic compounds and the formation of multiple phases during the dehydrogenation [154]. Thus, efficient regeneration procedures are still in development to further develop this technology. Complex hydrides can be grouped depending on their metallic anion. Alanates like NaAlH4 [155, 156] (7.4 wt%H2 and 67 gH2/L) and borohydrides like LiBH4 [157] (18.5 wt.%H2 and 122 g H2/L) are the most studied complex hydrides, but new amide-hydride composites like Li(NH2)–2LiH [158] (10 wt.%H2 and 104 gH2/L) are presenting promising properties.
4.2.3 B–N H2 carriers
B–N hydrogen carriers are composed of two key atoms: boron which acts as a metallic center for hydrides and nitrogen for protons. Ammonia–borane (AB) was initially proposed as an alternative to borohydrides due to its high gravimetric and volumetric densities (19.4 wt.%H2 and 144 gH2/L, resp.) [159]. As AB is a solid, liquid phase dehydrogenation of AB is carried out in a protic solvent like water or methanol. However, this hydrolysis or alcoholysis induces the formation of oxidized dehydrogenated boron species that require a harsh regeneration with the extensive use of strong hydrides or other complex processes [160, 161]. The dehydrogenation of solid AB can be achieved by thermolysis. Heating up to 100 °C is sufficient to dehydrogenate AB to linear, branched, or cyclic polyaminoborane species depending on the reaction environment (5) [162]. Two other H2 equivalents can be produced when increasing the temperature to 120–130 °C and 500 °C in order to form polymeric borazine and Boron Nitride (BN) as presented in equations (6) and (7) respectively [163, 164].![Mathematical equation: $$ n\mathrm{B}{\mathrm{H}}_3-\mathrm{N}{\mathrm{H}}_3\to {\left[-\mathrm{B}{\mathrm{H}}_2-\mathrm{N}{\mathrm{H}}_2-\right]}_{\mathrm{n}}+n{\mathrm{H}}_2, $$](/articles/stet/full_html/2023/01/stet20230038/stet20230038-eq5.gif) (5)
(5)
![Mathematical equation: $$ {\left[-\mathrm{B}{\mathrm{H}}_2-\mathrm{N}{\mathrm{H}}_2-\right]}_n\to {\left[-\mathrm{B}{\mathrm{H}}_2=\mathrm{N}{\mathrm{H}}_2-\right]}_n+n{\mathrm{H}}_2, $$](/articles/stet/full_html/2023/01/stet20230038/stet20230038-eq6.gif) (6)
(6)
![Mathematical equation: $$ {\left[-\mathrm{B}{\mathrm{H}}_2=\mathrm{N}{\mathrm{H}}_2-\right]}_n\to {\left[\mathrm{BN}\right]}_n+n{\mathrm{H}}_2. $$](/articles/stet/full_html/2023/01/stet20230038/stet20230038-eq7.gif) (7)
(7)
As BN is a very stable chemical, the dehydrogenation is usually limited to the first two steps to ensure the regeneration of the material, diminishing the effective densities to 12.9 wt.%H2 and 96 gH2/L. In addition, side-products like ammonia and other boron and nitrogen-containing gaseous products have been reported in large quantities (>20 wt.% of the AB weight) due to AB decomposition [165]. However, as the mechanism of degradation is heavily dependent on the conditions of the reaction (solvent, solid-state, etc.), it is not yet fully elucidated. AB solubilization in an aprotic solvent like ionic liquids improved the dehydrogenation as the solvent disrupted the proton-hydride intramolecular bonding and limited the carrier oxidation [166]. Similarly, dopant addition diminished the induction period and increased the kinetics and selectivity of dehydrogenation [167]. Nanoconfinement of AB in a nanostructure acted in the same fashion as metal hydrides (see 4.2.2.2) [168]. Lastly, the incorporation of alkali, alkaline-earth, or metals (Al) to the AB structure created heavier amidoboranes, where the partial replacement of the protons by a more electropositive element increased the selectivity and dehydrogenation kinetics at the cost of a portion of the H2 capacity [169–171].
4.2.4 Circular hydrogen carriers
Circular hydrogen carriers store H2 through chemical bonds on small gaseous molecules like N2 or CO2 to form respectively ammonia (NH3) or methanol (MeOH) (Fig. 11). Other products like hydrazine, formic acid, formaldehyde, methane, dimethylethers, carbonates, or carbamates are also circular hydrogen carriers but they will not be discussed due to their comparatively low technological development.
The main advantage of circular H2 carriers is their convenient transportation properties that allow for the production of H2 at a place of convenience where the gaseous lean carrier can be captured before recycling. In addition, the hydrogenated carriers are also high-value chemicals that can directly be used in chemical processes but H2 will not be retrieved in that case. The main issue of this technology revolves around the necessary gas separation and purification of the lean carriers from H2 during dehydrogenation as well as large-scale gas handling.
4.2.4.1 Ammonia
Ammonia is a valuable chemical for the synthesis of fertilizers that can act as an energy vector able to store H2 in liquid form with good gravimetric (17.8 wt.%H2) and volumetric (107 gH2/L) densities when pressurized at 8.6 bar at room temperature. Current production is guaranteed by the Haber–Bosch process which produces roughly 185 million tons of NH3 per year in 2020 [173, 174].
-
Hydrogenation
NH3 is produced in extreme conditions of temperature and pressure from N2 and H2 with a low conversion of 15% per pass, thus requiring numerous cycles of the reactants to achieve complete conversion (8).
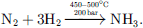 (8)
(8)
In addition, as presented previously (see Sect. 3), most of the currently produced H2 originates from fossil fuels. As a consequence, this industrial process consumes between 1 and 2% of the annual global energy production and generates 3 equivalents of CO2 per 8 equivalents of NH3 [175, 176]. Nevertheless, as this process is almost completely optimized, CO2 mitigation can only happen by developing low-carbon H2 sources (water electrolysis instead of SMR) and by implementing CCS. New ammonia production processes are also studied in order to create an improved disrupting process. Current alternatives require high electrical input and/or temperatures to produce ammonia via electrochemistry, either directly from N2 with water, in a two-step process where N2 reacts with H2 produced by electrolysis or a Li-mediated three-step cycle where the hydrolysis of Li3N facilitates the production of NH3 [177, 178]. However, these new processes are still energy-intensive and their scalability is yet to be demonstrated.
-
Dehydrogenation
Ammonia decomposition was historically achieved at high temperatures (>400 °C) on Fe or Ru transition metal catalysts supported on alumina due to the endothermicity of the process (9) [179, 180].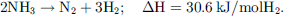 (9)
(9)
The decomposition rate is determined by the desorption of N2 that is dependent on the active metal (Ru > Ni > Rh > Co > Ir > Fe ≈ Pt > Cr > Pd > Cu ≈ Te, Se, Pb on Al2O3), the support (basic and conductor like Al2O3, MgO, CNT, …) and promoters (electron donors on Ru like K > Na > Li > Ce > Ba > La > Ca > pristine) [181–183]. NH3 shows promises as a H2 carrier, but its implementation for onboard applications is limited by the incompatibilities between NH3 and the PEM-fuel cell (PEM-FC) (high decomposition temperature, membrane poisoning, catalyst cost, NH3 toxicity and corrosivity) [184]. However, these NH3-PEM-FC incompatibilities can be partially lifted with an SOE-Fuel Cell (SOE-FC) [185]. Finally, NH3’s most practical application could be massive energy transportation to transport energy on an intercontinental scale.
4.2.4.2 Methanol
Methanol is one of the most produced chemicals worldwide with up to 164 million tons per year as a number of key industrial processes rely on it to produce high-value chemicals such as formic acid, formaldehyde, esters, ethers, olefins, and others [186–189]. From an H2 storage perspective, MeOH has attracted a lot of attention owing to its excellent gravimetric density of 12.5 wt.%H2 and volumetric density of 99 gH2/L. In addition, due to its only carbon, β-elimination cannot take place, limiting the number of potential side reactions.
-
Hydrogenation
MeOH formation from either CO (10) or CO2 (11) with Cu-based catalysts at high temperatures (300–450 °C) has been reported and patented many times since the 1960s [190–194].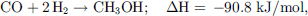 (10)
(10)
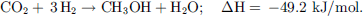 (11)
(11)
However, the harsh temperature conditions used in these processes furthered the research of processes with milder conditions. Nowadays, new production processes of MeOH from CO2 are emerging, like biogenic synthesis [195], amine/alkaline- [196] or acid-assisted [197] CO2 reaction, formic acid disproportionation [198], and gas–solid phase [199], but the scalability has yet to be demonstrated.
-
Dehydrogenation
MeOH non-oxidative dehydrogenation is a multi-step process that requires the presence of water. A first H2 equivalent can be retrieved by its dehydrogenation to formaldehyde (12), then, after the addition of water, 2 other H2 equivalents can be recovered to produce CO2 by the equations (13) and (14), yielding the overall equation (15).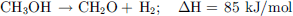 (12)
(12)
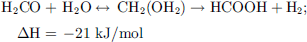 (13)
(13)
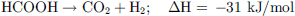 (14)
(14)
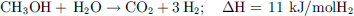 (15)
(15)
The dehydrogenation can catalyzed by heterogeneous catalysts based on Co, Ni, Cu, Pd, Ru, or Ir at temperatures superior to 200 °C [200]. A careful monitoring of the reaction conditions is necessary as the by-production of CO during the dehydrogenation could prove fatal to PEM-FC [201]. Despite its good compatibility with the current infrastructures and good biodegradability, MeOH has still a few barriers to overcome as its boiling point is low (64.7 °C) and its flammability and toxicity could prove too dangerous for public usage [202].
4.2.5 Liquid Organic Hydrogen Carrier (LOHC)
The Liquid Organic Hydrogen Carrier (LOHC) technology was developed in the 1970s to store the excess of nuclear electricity via water electrolysis in the form of automotive fuel [203, 204]. Recent developments and environmental considerations promoted this technology as a mean to store vast quantities of energy (GWh to TWh ranges) for a long time duration (seasonal). Much like the circular hydrogen carriers, the LOHC are liquid molecules able to store and release H2 at a desired place and time by catalytic hydrogenation and dehydrogenation reactions to respectively load and unload H2 (Fig. 12) [205]. However, both forms are liquid, which facilitates the separation of H2 and the LOHC during the dehydrogenation.
Storing H2 by covalent bonds on an organic liquid improves the handling and safety of the energy sector and retains similar volumetric densities compared to traditional physical systems, circumventing the need for heavy gas tanks and other cooling devices [206, 207]. The liquid phase is also advantageous as the reaction can proceed without the dilution of the H2 carrier in a solvent. In addition, most LOHCs are oil-like structures that can be easily transported by the current oil and gas infrastructures with small modifications to the system, eventually diminishing the implementation cost of the technology. Finally, social acceptance of the LOHC as a mobility option is favorable as the carrier system is similar to currently employed fuels [208].
Different criteria have been proposed to assess the potential of a LOHC. The energy efficiency is often addressed through the dehydrogenation enthalpy value. Classic LOHC systems have comparatively high enthalpy values (between 50 and 75 kJ/molH2) while the US Department of Energy (DOE) aims for 30–44 kJ/molH2 so that the H2 equilibrium pressure reaches 1 bar between −40 °C to 60 °C [209]. Like the other H2 storage systems, the gravimetric and volumetric energy densities highlight the energy storage efficiency with ultimate targets of 6.5 wt.%H2 and 50 gH2/L for a complete on-board system respectively. An excellent stability (>99.9%) of the LOHC is also required in order to avoid the replacement of the carrier [210, 211]. Finally, the LOHC must also answer to other application-dependent criteria such as a high liquid temperature range, its availability or ease to mass-produce from preferentially renewable feedstock, its cost, the H2 gas flow and quality on release (especially for fuel cells), the carrier toxicity and biodegradability [212]. It is worth noting that the properties of the LOHC can be heavily modified depending on the application and the time scale of storage. For example, a mobility application for individual transportation vehicles would mainly require high gravimetric and volumetric densities as well as a good cycling capacity and low dehydrogenation temperature while stationary systems (off-grid system) would be principally driven by the cost of the LOHC and its catalysts. Lastly, massive energy storage (seasonal storage) or a global energy system would necessitate both a low system cost and high densities.
The LOHC system possesses numerous advantages, but also drawbacks compared to other technologies. One major bottleneck is the toxicity of the LOHC molecules, similar to that of currently employed fossil fuels. Moreover, the classic LOHC systems are oil derivatives, so the development of bio-based structures would be preferential to completely remove fossil feedstocks from the LOHC technology. Besides, the catalysts used for the hydrogenation and dehydrogenation reactions are generally based on Platinum Group Metals (PGM) that are rare and expensive. Finally, the stability of the LOHC structures is a major concern, especially after multiple hydrogenation/dehydrogenation cycles with multiple side reactions occurring such as hydrogenolysis, cracking, and isomerization.
The next part of this literature review will focus on key molecules for the LOHC technology and the development of the associated heterogeneous catalytic systems and conditions for both the hydrogenation and dehydrogenation reactions. In addition, chemical functions able to store and release H2 will also be reviewed in order to broaden the perspective of LOHC structures.
To this date, no normalized procedure has been developed yet to compare reactions performed in different conditions like the temperature, pressure, reaction atmosphere, catalytic loading and composition, reactor type, and so on. In consequence, as the performance of the reaction is highly dependent on these parameters, it is often difficult to draw an easily generalizable conclusion. Whenever possible, key points were summarized in the introduction and conclusion of each part.
Finally, we would like to report some recently published reviews on the topic [212–216].
4.2.5.1 Cycloalkanes
Cycloalkanes have been the first structures studied for the LOHC technology, as homocyclic structures are cheap and readily available due to their presence in the oil refining processes. Moreover, the dehydrogenation of homocyclic structures is facilitated by the aromatization and the specificity of the C–H bond-breaking over C–C bond-breaking. Thus, the development of selective catalytic systems has been reported since the 1910s with seminal work from Zelinsky [217, 218]. From a thermodynamic standpoint, most aromatic structures have dehydrogenation enthalpies in the 50–80 kJ/molH2 range. In addition, conjugation in polyaromatic systems diminishes the energy needed d/uring dehydrogenation, with the exception of anthracene-based systems that see their dehydrogenation enthalpy increase after three conjugated cycles (Fig. 13).
 |
Figure 13 Dehydrogenation enthalpies calculated by the PM3 semi-empirical method as a function of the number of fused rings [219]. |
Clar’s rule links the stability in fused polybenzoic structures to the number of its stable sextets [220, 221]. Thus the stability increases from anthracene-type to phenanthrene-type to pyrene-type structures (Fig. 14). Moreover, anthracene-type structures produce Clar structures with a unique sextet but an increasing number of benzene rings, inducing the destabilization of larger structures.
 |
Figure 14 Clar structures for 4 fused rings: anthracene-type (left), phenanthrene-type (middle), and pyrene-type (type). |
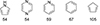 |
Figure 15 Influence of the size of the ring, the presence of N atoms, and their number on the enthalpy of dehydrogenation (kJ/molH2) [325]. |
Interestingly, graphene materials possess the best thermodynamic properties of polybenzylic structures, reaching theoretical dehydrogenation enthalpies in the 34–46 kJ/molH2 range [219]. However, due to their solid state, high fusion point, and low solubility, efficient hydrogenation and dehydrogenation of such materials are yet to be achieved [222, 223]. In addition, steric hindrance in fused ring systems is detrimental to the hydrogenation and dehydrogenation as reactivity limitations arise at the nods on the rings due to poor accessibility and the formation of less reactive isomers. In comparison, linearly linked hydrocarbons were shown to be more kinetically active [224].
Whilst numerous aromatic molecules have been tested for the LOHC technology, this work will focus on the most studied ones, i.e. the couples methylcyclohexane/toluene, decalin/naphthalene and perhydro-dibenzyltoluene/dibenzyltoluene. Benzene, benzyltoluene, fluorene, biphenyl and their respective hydrogenated counterparts will not be discussed due to the lack of significant system variations with the methylcyclohexane/toluene, perhydro-dibenzyltoluene/dibenzyltoluene, or decahydronaphthalene/decalin couples and the scarcity of development compared to the latter.
4.2.5.1.1 Methylcyclohexane/Toluene (MCH/Tol)
Due to its good gravimetric and volumetric densities (resp. 6.2 wt.%H2 and 48 gH2/L), abundance, low cost (0.3 €/kg), reactive simplicity, lower toxicity compared to the cyclohexane/benzene LOHC couple and lower computational cost compared to bigger LOHC like dibenzyltoluene, MCH/Tol is often used as a model carrier for the LOHC technology [213]. In addition, industrial development by the Chiyoda corporation has recently renewed interest in its research [225]. Recently, a conglomerate composed of Chiyoda, Mitsubishi, Mitsui, and the Japanese government demonstrated the feasibility of massive H2 transportation by ocean tankers with the MCH/Tol LOHC couple from Brunei to Japan.
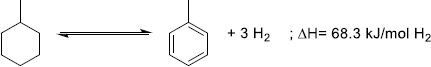 (16)
(16)
Historically, extensive research was conducted on the Methylcyclohexane–Toluene–Hydrogen (MTH) system for both mobile and stationary applications from the late 1970s to the late 1990s [205, 226]. Indeed, it was first proposed as an H2 fuel for automotive mobility in 1975 by Sultan and Shaw but was deemed inefficient compared to gasoline [203]. Taube and Taube proposed the MTH system again in the early 1980s as a solution to store excess nuclear power for automotive transportation [204]. Concomitantly, several pilot trucks were conceived to support the development of this technology [205, 227]. While the dehydrogenation reaction was not perfectly selective, this issue was circumvented by burning a fraction of the toluene or the impurities to kick-start and maintain the endothermal dehydrogenation. As the dehydrogenation is supposed to happen during times of low energy availability, lowering the energy consumption during the dehydrogenation is key to designing energy- and cost-efficient systems.
Further developments highlighted that the total catalytic oxidation of less than 10% of the produced Tol covered the complete calorie consumption of the dehydrogenation and a minimum of 6% could be in principle achieved by further optimization [228]. Similarly, extracting a portion of the combustion heat produced by an H2 thermal engine could compensate for the dehydrogenation enthalpy [229]. Moreover, this approach would be ineffective in the case of a PEM-FC engine due to its lower operating temperature. Finally, due to the low boiling point of MCH and Tol (resp. 100.9 °C and 110.6 °C), the dehydrogenation is a gas-phase reaction that is hence thermodynamically limited by H2 accumulation in closed systems [230]. Therefore, catalytic Pd–Ag membrane reactors were proposed as an answer to separate H2 from Tol during the reaction and push the thermodynamics forward [231]. Nevertheless, more development is required on these systems to reduce the amount of precious materials and to enhance their long-term stability in operating conditions [232].
-
Hydrogenation
Hydrogenation of cycloalkanes to aromatics was first achieved in 1911 and 1912 by Zelinsky with Pt and Pd catalysts supported on Al2O3 at 300 °C and 1 bar H2 [217, 218]. Since then, Ru/Al2O3 was shown to be the best monometallic catalyst for the hydrogenation of Tol to MCH, with extensive research on Pt, Ir and Rh based catalysts over the last three decades [233–235]. In addition, Ru presented a synergistic effect when doped with Pt [236]. Synergies were also studied for Pt, Pd, and Pd–Pt catalysts supported on Al2O3 to no avail [237]. However, alloying Pt and Pd increased the sulphur resistance of the catalyst at a temperature above 200 °C, while a rise of the support acidity increased the sulphur resistance at low temperatures (120 °C) [238].
Atmospheric pressure hydrogenation of Tol was also achieved by non-noble catalysts such as Ni/Al2O3 at 170 °C with perfect selectivity. Temperatures above 170 °C favored the desorption of H2, directly hindering the system kinetics [239]. Multimetal NiCoMo supported on zeolites allowed for the conversion of Tol to MCH at 200 °C and 20 bar H2. Side reactions such as ring contraction were observed in trace amounts [240]. Moreover, the hydrogen spillover effect, i.e. the migration of protons between the active metal nanoparticle and the support, also potentially played a role in the efficiency of the conversion. Indeed, hydrogenation with Pt/Al2O3 mixed/diluted with WO3/Al2O3 and solid acids showed improved conversion compared to the pristine catalyst due to the LOHC adsorption on acidic sites of the surface [241]. This effect was also observed on a Ru/NiCo/Ni(OH)2–Co(OH)2-nanoislands supported on carbon catalyst where the combination of the different sites achieved 100% conversion in liquid phase at 60 °C in 1 h for an activity ten times more superior to that of the equivalent Ru/C catalyst [242].
-
Dehydrogenation
Dehydrogenation of cycloalkanes to aromatics was also first achieved in 1911 and 1912 by Zelinsky with Pt and Pd catalysts supported on Al2O3 at 300 °C [217, 218]. However, these catalysts tended to deactivate due to preferential adsorption of the dehydrogenated products. Besides, side reactions like coke formation and dealkylation were observed at high temperatures (>350 °C) [243]. A series of noble metal catalysts supported on nitrogen-doped carbon showed that Pt was the most active catalyst, with the activity order Pt > Pd > Rh > Ir [244]. Extensive study of Pt supported on metal oxides and perovskites showed that 1 wt.% catalysts were more efficient than 3 wt.% catalysts, potentially due to the smaller size of the Pt nanoparticles [245]. Substrate modification with the addition of boron to a Pt/Al2O3 catalyst allowed for the tuning of strong acid sites into weak acidic sites, limiting the coke formation while retaining the stability of the metal nanoparticles on the substrate. This approach answered the lack of efficiency of alkaline addition that indistinctively covered all of the acidic sites and limited the dehydrogenation activity [246]. A Pt/Na–Y zeolite showed that hydrogenation or hydrogen transfer reactions happen during the dehydrogenation of methylcyclohexene, yielding both MCH and Tol [247]. As shown by previous contributions, the dehydrogenation rate-determining step was the desorption of toluene [248].
The synthesis of bimetallic catalysts is known to yield potential synergistic effects between the metals. Here, Pt–Re/Al2O3 showed a negligible influence of the H2 partial pressure on the dehydrogenation rate compared to Pt/Al2O3, indicating an alleviation of the thermodynamic equilibrium [249].
As the reduction of the active metal nanoparticle usually increases the activity of a catalyst, Single-Atom Catalysts (SAC) have a great potential to reach high kinetics. Recently, a Pt SAC supported on CeO2 nanorods was reported to catalyze both hydrogenation and dehydrogenation in continuous flow with activities 30 times superior compared to 2.5 nm Pt NP on CeO2 nanorods [250]. Similarly to Pt SAC, liquid metal solutions like Ga52Pt/SiO2 could atomically disperse Pt and maximize the gas-liquid interface, reaching up to 84.5% selectivity to Tol. While CO2 was observed, no coking formation was visible which was in agreement with the stable activity observed over 75 h [251]. Surface protonics are also a promising technique to lower the temperature of a reaction by applying an electric current [252]. Using a conductive Pt/TiO2 anatase catalyst submitted to a 5 mA electric current, the proton hopping specific properties of the support were increased, lowering the activation energy from 47.9 kJ/mol to 22.8 kJ/mol. Moreover, the dehydrogenation temperature could be decreased to 175 °C and the reaction equilibrium was displaced from 25% to 37% conversion [253]. As numerous effects occur simultaneously due to the specific nature of the TiO2 anatase support and electricity (Joule heating, etc.), more work is required to rationalize the effective influence of an electric field on dehydrogenation.
Non-noble metals are being heavily scrutinized as they may present an opportunity to replace critical and expensive PGM at usually the cost of higher catalytic loadings. The dehydrogenation of MCH to Tol was catalyzed by 20 wt.% Ni/Al2O3 catalyst, reaching a stable 92% conversion during 175 h on stream [254]. Further work on bimetallic Ni–M catalysts was pursued with Zn, Ag, Sn, and In. The Ni–Zn bimetallic catalyst showed a reduced activity (32.2% instead of 36.2% for pure Ni/Al2O3) with an increased selectivity to Tol (96.6% and 66.9% resp.). Nevertheless, the performance was still mediocre compared to a Pt/Al2O3 catalyst that reached full conversion and almost perfect selectivity (99.9%) in the same conditions [255]. Interestingly, flow kinetics with the NiZn catalyst in atmospheric pressure Ar demonstrated the insensitivity of the system to the MCH partial pressure above 0.1 bar and even a positive effect of H2 for partial pressure up to 0.4 bar, indicating that the hydrogenation of a species on the catalyst was necessary for the dehydrogenation. The increased selectivity by Zn doping was linked to the inhibition of low-coordination sites that were responsible for the C–C cleavage of the methyl group [256].
-
DFT
Computing techniques like Density Functional Theory (DFT) have been extensively used to rationalize and predict the reaction mechanism as well as propose catalyst modifications. A thesis on the dehydrogenation mechanisms of MCH on sub-nanometric Pt/Al2O3 proposed a DFT modeling coupled with kinetic experiments to rationalize the elementary steps during the dehydrogenation. DFT modeling showed that the cleavage of all C–H bonds was preferred compared to the migration and recombination of the protons. In addition, it exhibited the interactions between the LOHC and the support. Finally, the rate-determining step was difficult to assess due to the similar energies of either the third C–H bond cleavage or the desorption of Tol as often postulated in the literature [257, 258]. Predictive DFT was also used to estimate the influence of Sn-doping on a Pt/Al2O3 catalyst. The synthesis of an Sn4Pt phase could enhance the reaction kinetics due to lower activation energies compared to pristine Pt [259]. Lastly, DFT modeling showed that low-concentration of promoters (<1%) like Si, P or Se could in principle boost the dehydrogenation similarly to the already demonstrated S additives. Higher concentrations of doping elements showed no lowering of the reaction energy [260, 261].
-
Conclusion
From a techno-economic standpoint, dehydrogenation is the bottleneck of the MCH/Tol system as high-performance and low-cost PGM-free catalysts need to be developed while continuous H2 purification of the outlet feed and lower temperature of reaction are also to be addressed. In particular, the hydrogen performance, i.e. the stability of the LOHC, over the cycling is the principal concern due to isomerization and C–C cleavage side-reactions. Moreover, the high energy consumption during the dehydrogenation could be circumvented by using industrial heat waste or SOE-fuel cell systems. The rest of the economic chain is already ready as all dedicated infrastructures are in place and may just need a slight retrofit to adapt to the physical characteristics of the LOHC (higher viscosity) [262].
4.2.5.1.2 Decalin/naphthalene (Dec/Nap)
Decalin (Dec) is an inexpensive (0.6 €/kg) polycyclic fused-rings molecule that possesses high gravimetric and volumetric densities (7.4 wt.%H2 and 66 gH2/L resp.) [213]. However, naphthalene (Nap) is a solid up to 80 °C, which implies either incomplete conversion or dilution of the LOHC in a solvent in order to keep the system liquid. As calculated by Cooper, multiple fused rings diminish the enthalpy of dehydrogenation (Fig. 13) compared to free-standing or unfused rings [219, 263]. In addition, stereocenters are created by the fusion of the cycles, allowing for the formation of cis- and trans-hydrogenated isomers. Interestingly, the cis-isomer can undergo a ring flip reaction (17), while the trans-isomer has its conformation blocked and is the most stable isomer (18).
 (17)
(17)
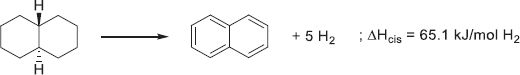 (18)
(18)
Similarly to the MCH/Tol couple, the boiling temperatures of decalin (185 °C) and naphthalene (218 °C) are lower than the reaction temperatures for both hydrogenation and dehydrogenation, implying either gas-separation and/or purification or thermodynamical limitations on the conversion in batch systems. In addition, a stable intermediate, tetralin (Tet), can form during the reaction (19).
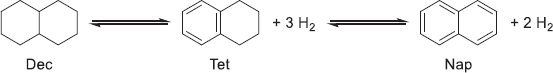 (19)
(19)
-
Hydrogenation
As Nap is a homocyclic LOHC, its hydrogenation to Dec can usually be achieved by noble metal catalysts such as Ru, Pt, or Pd at high temperatures (>200 °C) [264, 265]. However, control over which isomer forms preferentially is dependent on the catalyst, the reaction temperature, and the pressure [266]. The effect of the support was shown by a series of Pt–Pd supported on (Al2O3)(1-x)/(CeO2)(x) (x from 0 to 0.5) catalysts used for the hydrogenation at 290 °C and 55 bar in a batch reactor. The presence of CeO2 improved the conversion up to 99.5% in 3 h and this effect was attributed to the adsorption of Nap on acidic sites (Ce4+) as well as modification of the redox properties of the acidic sites and additional spillover reaction [267].
Non-noble catalysts such as NiMo supported on Zr, Al, or Ti-Hexagonal Mesoporous Silica (HMS) showed a higher selectivity to Dec due to the modification of the Mo=O or Ni oxide active sites by the Zr, Al, or Ti dopants while the less active NiMo/Al2O3 converted Nap to the intermediate Tet [268].
-
Dehydrogenation
Nap dehydrogenation happens in the gas phase due to the necessary high dehydrogenation temperature. To circumvent that issue, liquid film state reactors were proposed as they favor reactive distillation that is known to facilitate dehydrogenation compared to “suspended-state” conditions due to the constant removal of the dehydrogenated products [269]. These conditions could be achieved either by batch reaction in a large volume [270] or by spray pulse reaction [271]. Unsurprisingly, Pt was also the best metal to catalyze the complete dehydrogenation to Nap [272]. Experimental dehydrogenation on Pt/C and Pd/C showed that Pt was more active for Dec formation while Pd was more active for Tet formation in the same conditions. These results were rationalized by DFT, showing that molecular alignment on the metallic surface was more important than the intrinsic activity of the metals [273]. For the time being, only Pt with additives such as W and Re achieved a conversion superior to 90% in 1 h at 280 °C. Moreover, this system fulfilled the required H2 rate of fuel cells for mobility applications (50 kW, roughly 0.47 mol H2/s for a fuel-cell efficiency of 45%). By integrating the amount of precious material in the catalyst, the H2 release rate for Dec was 0.88 gH2/gPt/min [202, 274].
In order to improve this rate, the synthesis of highly dispersed Pt nanoparticles was undertaken. A relationship was found between a small particle size and a high specific surface of the support [275], the preparation method of the catalyst with ion exchange, polyol method, and chemical reduction being more efficient than precipitation and impregnation [276] and the presence of additives such as Ti or Ca during the synthesis [277]. Acid-neutralizing additives also limited the formation of coke promoted by strong acidic sites of the support [270, 277].
Pt supported on carbon materials was shown to yield excellent results (85% conversion and 95% selectivity to Nap, 5% to Tet) in contrast to catalysts supported on Al2O3, potentially due to the spillover of hydrogen on the activated carbon support [278]. The microstructure of the carbon support had a significant impact on the dehydrogenation properties of Pt as shown in Carbon Nanofibers (CNF): a positive effect was observed in the order platelet > fishbone > tube where platelet corresponds to the edges of stacked graphene and tube corresponds to basal graphene [279]. These results were confirmed by DFT calculations that showed stronger interactions between Pt and the edge planes of the CNF compared to the basal plane of CNT [280]. In addition, the wettability, i.e. the affinity of the LOHC with the support, influenced the catalytic activity of the system. Pt supported on CNT synthesized with O modifications induced a better Pt dispersion but a worse performance due to the lower affinity between the apolar LOHC and the polar surface [279]. On the contrary, N modifications doubled the catalytic activity compared to pristine CNT as N-modified CNT retained a good surface polarity and favored electron transfers from the metal to the support, facilitating the Nap desorption [281]. Electron transfers were also observed for a Pt/MgAl2O4 catalyst, linking the positively charged Pt particles to the weakened adsorption of Nap. In addition, the edges of the Pt nanoparticles were identified as the active sites of the catalyst by DFT and size analysis experiments [282, 283].
The presence of two hydrogenated isomers induced interesting kinetics effects. Indeed, the cis-isomer reacted faster than the trans-isomer due to its flexible nature that can better accommodate the active site [284]. In addition, the cis-to-trans isomerization on the support acidic sites was favored compared to the trans-to-cis isomerization [274, 285]. As the cis-to-trans isomerization and the dehydrogenation were also kinetically controlled by the temperature, with isomerization being favored at low temperature and dehydrogenation at high temperature, the dehydrogenation of Dec to Nap was slowed down by the less reactive trans-isomer [286]. Unfortunately, no solution was found to improve the dehydrogenation kinetics in this aspect.
The partial or total replacement of noble metals by transition metals was sought since the end of the 1970s when a Ni–Mo oxides/Al2O3 (80–100%)–SiO2 (0–20%) yielded low conversion (up to 25%) at high temperature (370 °C). An adverse effect was observed when increasing the SiO2 content, perhaps due to the stronger acidity of the support [287]. Since then, Pt–Ni/C catalysts have been suggested due to the metal’s synergistic effect. Surface Hydrogen Energy Binding (HBE) values were used as a descriptor of the dehydrogenation activity of the catalyst. Indeed, the deposition order of the metals (i.e. which metal was on top of the other) was correlated to the dehydrogenation activity: As the Ni surface presents a higher HBE than Pt, the former catalytic surface is more active than the latter [288]. Purely non-noble catalysts like 8%Ni–2%Cu/C catalysts were used in a spray-pulsed system at 350 °C for an activity 10 times superior to other catalytic systems in spray-pulsed mode. However, the activity is still 4.5 to 6 times lower than noble systems in batch or flow reactors. The Cu addition supposedly suppressed the hydrogenolysis reaction (cleavage of the C–C bond by H2) and promoted the C–H bond breaking [289]. Finally, Ni–WC/C replaced Pt without side reactions and coking in a flow system. 93% conversion and 100% selectivity to Nap were achieved at 300 °C with a high catalytic stability over 22 h. The good catalytic activity of Ni–WC was corroborated by DFT: Ni–Pt and Ni–WC had close HBE values, implying that they should have similar catalytic activities [290].
-
Conclusion
Despite better dehydrogenation enthalpy and gravimetric and volumetric densities compared to the MCH/Tol LOHC couple, Dec/Nap presents an intrinsic limitation due to the formation of highly stable intermediate and two hydrogenated isomers that hinder the system during both hydrogenation and dehydrogenation. While the boiling points of both Dec and Nap are slightly higher than those of the MCH/Tol couple, the dehydrogenation still happens in the gas phase, which further limits the system. Finally, Nap is a solid at room temperature, inducing either incomplete conversion, temperature control of the reaction/storage vessel, or dilution in a solvent. Therefore, the Dec/Nap LOHC couple probably exhibits too many barriers to be efficiently implemented as a LOHC.
4.2.5.1.3 Perhydro-dibenzyltoluene/dibenzyltoluene (18H-DBT/DBT)
Dibenzyltoluene (DBT) and its hydrogenated counterpart (18H-DBT) were first proposed as an LOHC couple in the 2010s and tremendous work by the Wasserscheid group and their collaborators has been achieved since then [291]. Hydrogenious, a start-up created by former members of the Wassercheid group promoted, with the help of a German consortium, the LOHC technology centered around the 18H-DBT/DBT couple [208]. 18H-DBT possesses good gravimetric and volumetric densities (6.2 wt.%H2 and 57 gH2/L resp.), while both forms are liquid on a wide range of temperature (−39 to 390 °C), albeit viscosity would increase drastically at temperatures below 20 °C [291, 292]. From a chemical risk standpoint, the 18H-DBT/DBT couple has a much lower toxicity and ecotoxicity than MCH/Tol and a lower vapor pressure than Dec/Nap, making it a safe couple to handle. In contrast, the 18H-DBT/DBT couple has a higher dehydrogenation enthalpy (20) [291].
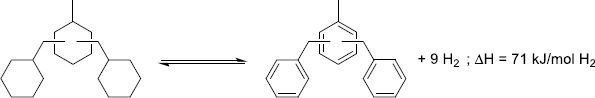 (20)
(20)
Finally, DBT is cheap (4 €/kg) and commercially available as a heat-transfer oil under the name Marlotherm® SH, owing to its excellent thermal stability [213, 293]. Scale-up projects for the DBT technology have started in both hydrogenation and dehydrogenation since 2017–2018 [294]. As extensive work on DBT-related systems was published in the literature, a part of this review will be dedicated to them.
-
Hydrogenation
Complete hydrogenation of DBT was first achieved by a Ru/Al2O3 catalyst in 4 h at 150 °C and 50 bar [291]. Recent advances proposed a K-doped Ru supported on MgO catalyst as K was shown by DFT to favor the heterolytic H2 adsorption at low concentrations by charge transfer from Ru to the substrate. At higher concentrations, K interacted directly with H2, decreasing the catalyst activity [295]. However, it also became clear that the hydrogenation occurred first on the side rings before the center ring, as shown by reaction monitoring with 1H NMR and High-Performance Liquid Chromatography (HPLC) [296]. As expected, the hydrogenation of the center ring was the rate-limiting step and the development of more active catalysts was undertaken. First, the complete hydrogenation of DBT by a Pt on Al2O3 eggshell was observed for temperatures above 220 °C and 30 bar [297]. Further work was carried out on the hydrogenation with Pt supported on Al2O3 at the reduced temperature of 140 °C and 30–40 bar in 35 min. The Al2O3 support was found more reactive than the SBA-15 (60 min), Hydroxyapatite (100 min), and activated carbon (280 min) supports [298].
Hydrogenation with non-noble metals was also achieved by a Ni Raney catalyst in 30 h at 170 °C and 50–70 bar [299]. Later, a Ni catalyst, NISAT 310, accomplished the complete hydrogenation at 150 °C and lower pressures (4–16 bar) in flow conditions [300]. Finally, a recent advance proposed the hydrogenation of DBT with a Mg2NiH4 metal hydride able to transfer H2 from the gas phase to the LOHC at 280 °C and 60 bar, reaching an adsorption of 5.70 wt.% (92% DoH) in 20 h [301].
-
Dehydrogenation
The dehydrogenation of 18H-DBT was first achieved with Pt- and Pd-based catalysts, with Pd catalysts being usually less active than their Pt counterparts. Support modification proved primordial, as activated carbon yielded the best results compared to Al2O3 and SiO2. In addition, the Pt loading lowering increased the dispersion of Pt on the surface and further improved the catalytic activity. Finally, temperature modification from 230 °C to 290 °C increased the conversion from 16% to 92% in 3.5 h, achieving a H2 dehydrogenation rate comparable to other state-of-the-art LOHC N-Ethylcarbazole (see para Perhydro-N-ethylcarbazole/N-ethylcarbazole (12H-NEC/NEC) from 4.2.5.2) [291]. Other studies confirmed the effectiveness of activated carbon as support and hinted that other metal oxides like TiO2 anatase-rutile nanopowder might compete with activated carbon, although further rationalization was still required [302]. While activated carbon was proven good support for Pt, the high reaction temperature (>300 °C) could prove detrimental to the stability of the catalyst. Thus, extensive work was pursued on more stable metal oxide supports like Al2O3 that achieved a PEM-FC-compatible H2 production rate of roughly 1 gH2/gPt/min [303].
The catalyst preparation method was also significant improving the activity, as a supercritical CO2 (sc-CO2) assisted Pt/Al2O3 catalyst was more active than its analog prepared by wet impregnation. However, this increased activity was not selective as more by-products and carbon impurities at the catalyst surface were observed for the sc-CO2 catalyst, potentially due to the catalyst’s higher surface acidity [304]. S-doped Pt/Al2O3 catalysts were prepared by solvent-deficient preparation in order to effectively block the defect sites that formed by-products on the Al2O3 surface. A better contact between Pt and S was also achieved compared to wet-impregnation, increasing the creation of electronically deficient species Pt that weakened DBT adsorption and favored 18H-DBT adsorption. Moreover, the catalyst was stable over five cycles without sulphur losses even during the regeneration of the catalyst under H2 [305]. Finally, the Glycine Nitrate Process (GNP) was also used to simultaneously synthesize by combustion the active metal and support, producing highly dispersed Pt/Al2O3 and Pt/CeO2 catalysts. Pt/CeO2 was more active than Pt/Al2O3, probably due to the smaller pore size of the Al2O3 support that limited the mass transport of the LOHC. Nonetheless, Pt/CeO2 was still four times more active than a benchmark Pt/Al2O3 catalyst (81% and 18% conversion in 2.5 h resp.) [306].
Recent advances revealed that the H2 bubble nucleation was a rate-limiting step under unfavorable mixing conditions. Indeed, H2 supersaturation limited the liquid–gas oscillation in the pores of the catalyst, effectively decreasing the Pt/Al2O3 catalyst activity to less than 1.8% of its original activity. Bubble nucleation inhibition was linked to the excellent wettability of DBT on the catalyst support and hydrophilisation of the support lifted the nucleation inhibition. However, poor wettability could also lead to reduced performance [279]. Catalyst drying to reintroduce gas–liquid interfaces and overheating of the catalyst pellet were also efficient methods to lift the nucleation barrier [307]. Extra work was pursued to increase the selectivity by activating the Al2O3 surface with H2 and O2 plasma. The modification of hydroxyl surface groups (with O2) and oxygen vacancies (with H2) improved the Pt dispersion. However, an improved stability of the catalyst with better long-term performance was observed only when the number of hydroxyl groups was increased. Hydroxyl surface groups were reported to promote H2 spillover and the concomitant increase of Pt(0) proportion to reduce the number of side reactions [308].
Alloying Pt with other noble metals was pursued to reduce the coking of the catalysts. However, Pt-Pd alloying was reported less active than Pt and Pd and these activities were linked to the H abstraction energies by DFT. In theory, specific atomic layering Pt–Pd–Pt could yield similar results to pristine Pt [303]. However, another DFT study proposed the Pt–Pd–Pd combination [309]. Finally, the DBT adsorption on Pt, Pd, and PtPd was also dependent on the catalytic surface lattice in the order (1 1 0) > (1 0 0) > (1 1 1) [310]. As DFT is heavily dependent on the calculation conditions (functional, basis set, convergence criteria, etc.), conclusions based on DFT articles are not universally applicable, and more experimental work would be required to confirm these results.
Alloying with transition metal was accomplished with a Pt–WOx/Al2O3 catalyst. When 22% of Pt was replaced by W, the catalyst presented better yields (+6 to 9%) which were attributed to a spillover effect of H to W confirmed by DFT calculations [311]. DFT studies also pointed at other transition metals like Cu, Ni, or Fe to tune the electronic properties of Pt. The rate-determining step was the first C–H bond breaking on the middle ring and the reaction energy was linked to the H adsorption energy [312]. Therefore, transition metal alloying might allow for a reduction in the amount of critical PGM in the catalytic system. Noble metal-free catalysts could also be an efficient PGM-reduction strategy, although no active catalyst to this date.
-
System
The 18H-DBT/DBT couple has been an excellent candidate to model and design LOHC systems whose physical, chemical, and thermodynamical properties are scarce. Thermophysical and chemical parameters of the DBT system were measured and calculated to favor the process design, modeling, and engineering of the LOHC couple [292]. While GC-FID and 13C NMR ex-situ follow-up techniques were classically used to assess the hydrogenation and dehydrogenation reaction progress as a function of the pressure and temperature of reaction [313], machine learning was recently proposed to predict the influence of the temperature, pressure, stirring speed and relative quantities of catalyst and DBT on the H2 storage. Up to 98.7% accuracy was achieved using data from the literature [314]. Moreover, online measurements of the physicochemical properties of the reaction mixture such as the density, refractive index, UV–Vis, Raman spectroscopy, and viscosity were proposed to replace the classic lab-scale GC, NMR, or elemental analysis for industrial application. Density or refractive index was highly promising due to the intrinsic measurement accuracy and the low-temperature dependence of the measurement as well as the small deviation in a measure even for different mixtures of same the DoDH (Degree of Dehydrogenation, i.e. the amount of evolved H2 with regard to the stored amount of H2 in the LOHC). Ultraviolet-visible spectroscopy (UV–Vis) was limited by the high absorbance of aromatic compounds that complicated the measurement process at higher concentrations of DBT. Raman was deemed challenging and costly for a low accuracy to follow the evolution of the LOHC. However, its low-temperature dependence and usefulness in probing other parameters of the system such as the catalyst could prove interesting. Finally, the viscosity measurements were inefficient as various mixtures with the same DoDH exhibited different viscosities in the case of the 18H-DBT/DBT couple [315].
Reactor development is key to fully optimizing the LOHC system. Dehydrogenation with Pt/Al2O3 in 12 h at 290 °C in a microchannel reactor allowed for better conversions than its stirred reactor counterpart (58% vs. 19% resp.), which permitted to use of less catalyst for equivalent performances [316]. The development of swing reactors and reversible catalysts like Pt/Al2O3 allowed for hydrogenation and dehydrogenation to occur in the same reactor. Thus, compact systems demanding less handling (no operation between the hydrogenation and dehydrogenation) with decreased cost (only one reactor) could be conceived. Moreover, such systems had a higher reactive availability as they could be kept heated up due to the hydrogenation and dehydrogenation being controlled only by the equilibrium pressure (around 3 bar for 18H-DBT/DBT) and the stirring speed. The catalyst was also regenerated at high pressure of H2 during the hydrogenation, keeping its activity over four cycles [297]. Nevertheless, another study confirmed the feasibility of a reversible reactor with Pt/Al2O3 but a rapid loss of cycling capacity (−25% after 1 cycle, hydrogen capacity decreased from 60% to 20% after 5 cycles) was observed and attributed to the deactivation of the catalyst due to coking, surface modification and side-reactions of DBT such as ring opening and cracking [298]. Thereby, reversible systems present an interesting option for stationary H2 generation and storage, providing their optimization for both reactions and connection to industrial waste heat to facilitate heat integration.
The LOHC stability is key to ensure the reuse of the structure over numerous cycles and its evolution was covered by multiple articles in the literature. Early work quantified only traces (<0.01%) of by-products during the dehydrogenation at 270 ° in 72 h [291]. The system resilience was later estimated to be 14,000 h under hydrogenation conditions (150 °C and 50 bar H2) while 8000 h were estimated under dehydrogenation conditions (310 °C and 1 bar) [317]. Dehydrogenation stress tests at 355 °C showed the formation of by-products such as benzyltoluene, toluene, xylene, methylfluorene, and their isomers in the liquid phase while CH4 was observed in the gas phase. The impurity’s disappearance after the hydrogenation implied that cracking reactions also happened on the hydrogenation catalyst. If the limitation for toxic byproducts is low (<0.5 mol%), DBT use would be limited to less than a year as a model built on accelerated stress tests predicted the formation of 7.4 mol% impurities after 89 h reaction in the normal operating conditions (300 °C) [300].
The H2 quality obtained from the DBT dehydrogenation must be stringent (>99.99%) as fuel cells cannot tolerate high contaminant levels, especially CO. The impurities in the produced H2 came from the impurities found initially in the DBT like water that produced CO and CO2 as shown by Infrared (IR) spectroscopy and isotopic replacement with O18. The addition of Dicyclohexylmethanol to represent oxidized organic species also favored the production of oxygenated impurities as a correlation was found between the organic alcohol and the CO and CH4 levels. By using a recycled LOHC whose impurities were already phased out or by purifying/drying the LOHC beforehand, the H2 stream quality improved. Both methods increased the H2 purity to >99.999% levels, with CO being found at traces level (<0.2 ppmv) [318]. Another technique usually reserved for gas phase dehydrogenation was the use of a PdAg membrane reactor that diminished the impurities in the gas phase from 200 ppmv in the reactor from 3 to 7 ppmv after the membrane. Nevertheless, contaminant traces could disturb the H2 flux in the membrane, making routine cleaning membrane procedures mandatory to ensure a continuous H2 flux quality [319]. Carbon filters also proved efficient in purifying contaminants in the H2 gas feed with a constant 20 ppm CH4 contamination and aromatic contamination increasing from 2 to 6 ppm over 9 h. The H2 output was controlled by using a simple pressure algorithm and a buffer volume, conveniently regulating the pressure variations due to the reactor temperature variations and allowing for an output-connected PEM-FC to produce 6.6 kW over 4.5 h in a dynamic system [320].
The modification of the LOHC properties was performed by mixing DBT with Benzyltoluene (BT) to lower its viscosity for applications in colder regions. The dehydrogenation was improved by 12–16% at 260 °C when compared to pure DBT due to the presence of BT in the gas phase that diluted the H2 in the gas-phase, thus displacing the reaction equilibrium similarly to reactive distillation [321].
Finally, 18H-DBT and the other hydrogenated LOHC were used as an H2 source for transfer hydrogen reactions. The reaction of 18H-DBT with Tol allowed for an almost thermoneutral reaction to produce MCH with a conversion superior to 99% in 5 h on a Pt/Al2O3 catalyst [210]. DBT was also used for the transfer hydrogenation of Acetone to Isopropanol in order to produce electricity with direct Isopropanol/Acetone fuel cells. Pt/SiO2 was reported as a great candidate for this system as SiO2 limited the formation of Acetone condensation by-products [322]. Direct Isopropanol fuel cells reached up to 254 mW/cm² at 0.55 V, comparable to Methanol/air fuel cells, demonstrating the interest in this technology [323, 324].
-
Conclusion
18H-DBT/DBT has been well developed from a catalytic standpoint and its system integration was also rigorously described in the literature. Most importantly, considerations such as online measurements, reactor design, LOHC stability, H2 quality and purification as well as physicochemical properties modification and alternative fuel cell development were covered. From a commercial perspective, DBT was extensively marketed by Hydrogenious and social acceptance of such LOHC carriers would be high due to its resemblance to a non-flammable oil. However, its high dehydrogenation enthalpy and temperature intrinsically limit the applicability of this LOHC if no free heat is provided.
4.2.5.2 N-heterocycles
In the 2000s, Cooper proposed N-heterocyclic from the carbazole family as LOHC due to their reduced enthalpies compared to their homocyclic analogs. Their study highlighted the high potential for hydrogenation and dehydrogenation of conjugated 5-membered rings fused with 6-membered ring structures [219]. DFT modeling by Clot et al. linked the integration of N atoms to the lowering of the dehydrogenation enthalpy depending on their number, position, and size of the ring (5 or 6) [325].
Further modeling showed that including electrodonating substituents stabilized the aromatic cycle, decreasing the enthalpy as a function of the Hammett parameter σ (para) [326]. Moreover, faster dehydrogenation kinetics were observed for heterocycles compared to the analogous homocycles due to the destabilization of the Cα–H bond induced by the N atom [327].
Whilst numerous heterocyclic molecules have been tested for the LOHC technology, this work will focus on the most studied couple dodecahydro-N-ethylcarbazole /N-ethylcarbazole (12H-NEC/NEC) as an example of a 5-membered N-heterocycle and the promising couple dodecahydro-phenanzine/phenazine (12H-PHE/PHE) as an example of 6-membered N-heterocycle. Pyrrole, indoline, pyridine, triazolidine, quinoline, naphthyridine, carbazole, their alkylated or arylated derivatives, and their hydrogenated counterparts will not be discussed due to the lack of significant system variations with the 12H-NEC/NEC or 12H-PHE/PHE couples, their poor stability, selectivity or low gravimetric density due to incomplete dehydrogenation.
4.2.5.2.1 Perhydro-N-ethylcarbazole/N-ethylcarbazole (12H-NEC/NEC)
Inductive and mesomeric donations as well as conjugation and aromaticity favor lower enthalpies, hence carbazoles, indoles, or their derivatives are attractive for the LOHC technology. Seminal work by Cooper demonstrated the capacity of this class of compounds to selectively store and unload H2 as well as their lower dehydrogenation enthalpy with regard to classic systems [219, 328]. Carbazole and their derivatives showed the best results out of the structures presented in Figure 16.
 |
Figure 16 Examples of structures studied by Cooper From left to right: carbazole, indolo[3,2,1-jk]carbazole, indole, pyrroloindole, bis-indolylmethane and pyrrocoline. R= H, alkyle [219]. |
Carbazole presents high gravimetric densities (6.7 wt.%H2), however, its high fusion point (250 °C) is detrimental as it cannot exist as a liquid at near ambient temperatures or must be diluted in a solvent. Alkylated analogs such as NEC have a much lower melting point (70 °C) which facilitates their use while still possessing good gravimetric and volumetric densities (5.7 wt.%H2 and 63 gH2/L resp.) with a reduced dehydrogenation enthalpy of 51 kJ/molH2 (21) [213, 219]. In addition, N-alkylation prevents catalyst poisoning and the subsequent slower catalytic activity due to the strong N adsorption on the metal nanoparticles [327]. Moreover, the advantage of the alkylation compared to the simple carbazole was demonstrated by DFT in particular for the dehydrogenation. Indeed, the interaction strength between the NEC, carbazole, fluorene, and a Pd surface was probed, revealing that carbazole interacted strongly with Pd by its N moiety comparatively to NEC, while fluorene had an even stronger interaction [329]. Experimentally, the presence of the ethyl group facilitated NEC desorption, thus freeing the active site and increasing the catalytic activity [330].
 (21)
(21)
NEC has been extensively studied as an LOHC material since its discovery by Air Products and new LOHC players in Asia have been rising since 2014. Hynertech was founded by a former Air Products partner, Prof. Hansong Cheng, and chosen by the Chinese government to develop energy storage technologies at the destination of the Chinese market in order to cover logistics mobility applications [331]. The first 10,000 tons production plant of Hynertech LOHC was operational in 2020 and, in November 2022, an H2 storage and supply system releasing 400 kg H2 per day was commissioned at a price of 40 €/kg [213, 332].
-
Hydrogenation
NEC hydrogenation was first performed in diluted THF solution or simply melted with Ru, Rh, and Ni Raney catalysts. Ni Raney 2800 was particularly efficient as it allowed for the in-situ hydrogenation and synthesis of NEC from carbazole and the desired anhydrous alcohol (22) [219].
 (22)
(22)
Another study with Raney-Ni showed the effect of temperature and pressure on the direct hydrogenation of NEC to 12H-NEC, with an optimum at 200 °C and 50 bar H2 respectively, completely carrying out the hydrogenation in 1 h [333]. NEC hydrogenation was also performed with Ru/Al2O3 in 1 h at 150 °C and 70 bar H2, yielding 98% conversion and a selectivity superior to 95% for 12H-NEC [330]. Similarly, Ru/Al2O3 yielded 100% conversion and 98% selectivity to 12H-NEC in 1 h at 140 °C and 60 bar, with a stringent effect of the H2 pressure, stirring speed, and catalyst dosage [334].
Both commercial and synthesized by chemical reduction catalysts were screened, showing that the active metal reactivity order was Ru > Pd > Pt > Ni and the support order was Al2O3 > TiO2 > SiO2–Al2O3 > zeolite > graphite > activated carbon. The synthesized catalysts yielded better results due to better atom efficiency: an inverse relationship between the catalytic activity and the particle size was observed [335]. Subsequent hydrogenation studies at 130 °C and 70 bar confirmed the active metal reactivity order with Ru > Rh > Pd and linked the catalyst activity to the d-band center position. Indeed, a low d-band center indicated a strong interaction between the metal and the NEC, implying that the rate-determining step was the adsorption of NEC on the surface [336]. However, a DFT study proposed the 10H-NEC to 12H-NEC conversion as the rate-determining step which was confirmed by a later study [337, 338]. However, as the DFT study was applied only to the LOHC structure and not to the LOHC-metal system, energy levels might vary considering the various stable intermediates forming depending on the metal: 8H-NEC on Ru, 4H-NEC on Pd, and no stable intermediate on Pt. The hexagonal compact packing structure of Ru, compared to the face-centered cubic structure of other metal surfaces was also linked to the stability of 8H-NEC [339].
Moreover, 12H-NEC isomerization was observed. As 12H-NEC possesses 4 stereocenters due to the presence of fused rings, 16 potential isomers exist, while only 6 isomers are possible due to symmetries. The trans, trans isomer (A) is the most stable, and the cis-syn-cis the least stable (B) (Fig. 17).
 |
Figure 17 Isomer structures of the 12H-NEC. The most stable is the isomer A, while the least stable is the isomer B. The structures A to D are symmetric while the structures E and F are asymmetric. |
The production of unstable isomers could in principle simplify the dehydrogenation as less energy would be required [219]. Both symmetric and asymmetric products were observed by 1D and 2D NMR techniques by using the number of carbon resonances to determine the symmetry (8 for symmetric compounds and 14 for asymmetric compounds in 13C NMR) [340]. As the H2 addition was supposedly concerted, asymmetric products imply that isomerization occurred either due to a NEC-metal desorption-readsorption mechanism on weakly bonding metal particles or due to the hydrophilicity of the support [336, 339]. DFT on flat Ru (0 0 1) and low-coordination sites (1 0 9) confirmed that isomers formed from the desorption of the 8H-NEC from flat (0 0 1) due to steric constraints and readsorption to edge sites (1 0 9), limiting the activity of the Ru catalyst as a side-effect [335]. Finally, DFT, Conductor-like Screening Model (COSMO), and Molecular Dynamics (MD) simulations corroborated that unstable isomers were favored at low temperatures due to higher energy efficiencies [341].
Bimetallic catalysts like Pd2Ru/SiCN also yielded better results than the sum of their part or Ir and the corresponding commercial Al2O3 or C supports [342]. Further development aimed at the reduction of precious metal in the catalyst composition. Therefore, Ru–Ni catalysts were synthesized on TiO2 whose structure was primordial. Rutile was more active than anatase, but less selective while a commercial anatase/rutile in a 1:4 mix yielded improved results compared to both pristine phases due to the formation of smaller Ru NP on this support. Finally, Ni addition to Ru slightly increased the activity compared to Ru due to a potential hydrogen spillover effect [343]. A spillover effect was also reported for a Ru4.5Ni0.5 supported on a biochar catalyst prepared by carbothermic reduction. Better catalytic activities and stability were obtained compared to chemically reduced catalysts due to partial graphitization of the surface that favored electronic transfers and the embedding of the active Nanoparticles (NP) in the support cavities, maintaining similar performances over 10 cycles [344]. Lately, a Ru0.7Ni0.3/SBA-15 (mesoporous silica) catalyst yielded better hydrogenation performances at 60 °C than a commercial Ru/Al2O3 catalyst at 90 °C due to improved electronic transfers from Ni to Ru. The good stability between the NP and the support was attributed to the ionic interactions with the hydroxyl surface groups [345]. Similar results were also observed on a Ru/Layered MgAl Double Hydroxide (LDH)/CNT prepared by ultrasonication that rapidly evolved 98.4% H2 in 24 min at 120 °C and 60 bar and was stable for 8 cycles. The improved catalyst was also more active at 80 °C than a classic Ru/Al2O3 due to highly dispersed Ru NP and fast electron transfers between the LDH and the CNT [346].
Hydride additions to the catalyst as a support material for Ru yielded better performances than a classic Ru/Al2O3 catalyst. YH3 was comparatively more efficient than LaH3 and GdH3 due to hydride transfers from the support to the adsorbed Ru-NEC materials and the regeneration of hydrogen-deficient YH2-x to YH3 by H2 in solution, allowing for complete hydrogenation at 90 °C and 10 bar [347]. These results were confirmed on a Ni-YH3/Al2O3 catalyst that achieved complete conversion when both YH3 and Ni/Al2O3 showed no activity. The mechanism was unchanged with H transfer from YH3 to the Ni-NEC interfaces and YH3 as an H2-splitting site. The contact between Ni and YH3 was primordial as 500 nm Ni particles were more reactive than 10 nm Ni nanoparticles [348]. A composite Co-B-YH3/Al2O3 catalyst also showed a remarkable activity comparable to the best Ru catalysts. However, a gradual reduction of activity was observed over 5 cycles due to the phase detachment from the support [349]. Finally, LaNi5.5 nanoparticles, or LaNi5 core with a Ni-rich shell, catalyzed the complete H2 absorption in 8 h at 180 °C, and 70 bar H2. The LaNi5 phase could form a partial hydride phase depending on the pressure that enabled H transfers to the Ni shell. While no H2 capacity modification was noted, a kinetics decrease was observed after 9 cycles due to the partial decomposition of the LaNi5 to LaH3 [350].
-
Dehydrogenation
Seminal work by Cooper described the dehydrogenation reactivity of the active metal in the order Pd>Pt>Ni over alumina and lithium aluminate, with Pd/Al2O3 reaching the best conversion. Re–doped Pt catalyzed the dehydrogenation comparatively to Pd/Al2O3, while Pt–Pd or Pt–Sn catalysts were less active. The addition of 3 mol% additives had a neutral effect for Brønsted acids and bases and a negative effect for Lewis acids or bases. Finally, H2 purity measurements revealed that low methane formation (<200 ppm) was observed for both Pt–Re/Al2O3 and Pd/Al2O3 while ethane formation due to the decomposition or dealkylation of NEC was detected in concentrations superior to 6000 ppm for Pt-Re/Al2O3 and below 50 ppm for Pd/Al2O3 [219].
From an activity standpoint, the H2 production rate was extremely dependent on the temperature. At 270 °C, up to 9 gH2/gPt/min were observed for a Pt/Al2O3 catalyst for a DoDH of 100%. However, thermal stability tests revealed roughly 2% degradation due to dealkylation after 72 h [291]. By diminishing the temperature at 180 °C, the activity decreased to 0.67 gH2/gPt/min with the same catalyst at DoDH = 100%). Finally, Pd/Al2O3 was more active and selective than Pt/Al2O3, reaching 0.8 gH2/gPd/min at DoDH = 100%, equivalent to the best DBT performances [351].
The dehydrogenation mechanism with Pd/Al2O3 followed a three-step process, starting from the center ring and with the formation of the 8H-NEC and 4H-NEC intermediates. Interestingly, each dehydrogenation step started at a different temperature (128 °C, 145 °C and 178 °C for the conversion of the 12H-NEC, 8H-NEC, and 4H-NEC species respectively), indicating that the temperature dehydrogenation could not be reduced below 178 °C to achieve completion. Moreover, the LOHC could be cycled 10 times with less than 0.1 wt.% capacity loss and a high H2 purity of >99.99% with the only gaseous by-product being ethane [352]. Advanced analytic studies on Pd/Al2O3 model catalysts in Ultra-High Vacuum (UHV) with Infrared Reflection Absorption Spectroscopy (IR-AS), DFT, and High-Resolution X-ray Photoelectron Spectroscopy (HR-XPS) showed the adsorption of 12H-NEC on both the metal NP and the support with preferential migration to Pd [353]. The adsorption of 12H-NEC started first from −100 °C, forming only a monolayer by desorption of non-chemisorbed species from −63 °C. The C–H activation of the 5-membered ring started from −50 °C and the 6-membered ring from 0 °C. C–N scission was observed above 77 °C [354]. Similar work was carried out on Pt(1 1 1) with in-situ synchrotron radiation-based HR-XPS. The dehydrogenation in UHV presented similar patterns: adsorption of multi-layers of 12H-NEC from −133 °C, desorption of the physisorbed 12H-NEC to let only a mono layered chemisorbed 12H-NEC up to −33 °C, dehydrogenation to 8H-NEC (57 °C), then to NEC at 107 °C before carrier degradation above 117 °C to form Carbazole or carbon residues on the catalyst. The dehydrogenation started with the formation of the pyrrole ring before the benzene ring which required higher temperatures [355]. The same results were observed in UHV with IR-AS, DFT, and classic HR-XPS [356]. Further studies indicated that the Pt edge and corner sites favored C–N bond scission at low temperatures and that well-ordered Pt facets favored dehydrogenation. Therefore, bigger particles would be required, consequently diminishing the reactivity [357].
Numerous kinetics studies on various supports such as Al2O3 [351], SiCN [342], TiO2 [358, 359], reduced Graphene Oxide (rGO) [360], and SiO2 [330, 361] revealed that the metal reactivity order was usually Pd > Pt > Rh ≈ Ru > Au. For most Pd catalysts, the rate-determining step is the 4H-NEC to NEC dehydrogenation, with the kinetics constants diminishing in the order 12H-NEC to 8H-NEC > 8H-NEC to 4H-NEC > 4H-NEC to NEC [351, 359–364]. These results were confirmed by DFT [365]. Only one study reported the 12H-NEC to 8H-NEC dehydrogenation as the rate-determining step for Pd supported on Al2O3, TiO2, and SiO2, except for the activated carbon support where the 8H-NEC to 4H-NEC dehydrogenation was [366]. The effect of the support on Pd catalysts was probed by different studies, with the classic reactivity order being SiCN > C > Al2O3 > TiO2 > SiO2 [342, 366]. However, the dehydrogenation process on Pd/Al2O3 was superior to Pd/C due to overall better kinetics that limited the accumulation of intermediates. Moreover, a volcano plot was observed when comparing the Pd NP size with regard to the catalytic activity, in agreement with previous studies [357, 366]. A high Pd reduction also yielded better results [366]. Finally, the facet-dependent activity of the dehydrogenation was proved by a series of Pd/rGO catalysts with (1 0 0), (1 1 0) and (1 1 1) facets. At 180 °C, the complete dehydrogenation was achieved in 30 min on the (1 0 0) facet, 2 h on (1 1 0) and 7 h on (1 1 1). This activity change was linked to better NEC adsorption on low-index facets. In particular, the Pd (1 0 0) facet facilitated the rate-determining step 4H-NEC to NEC and DFT characterized the rate-determining step as the conversion of 3H-NEC* to 2H-NEC* [365].
Highly ordered mesoporous supports have been the focus of recent advances. Pd supported on a MIL-101 MOF presented a better activity than commercial Pd/Al2O3, evolving 100% H2 in 4 h at 170 °C. 89% catalytic activity was retained after 5 cycles [363]. Moreover, a Pd/SBA-15 catalyst showed a remarkable 98.7% DoDH in 1 h at 180°C with high stability due to the ionic Pd–hydroxyl group [367]. Finally, Pd/MgAl-LDH synthesized by ultrasonic reduction achieved 100% conversion and 99.3% DoH at 180 °C in 6 h. 98% stability was observed after 6 cycles, compared to 86% stability after 3 cycles for an analogous chemically reduced catalyst due to sintering [368].
While less active and selective than Pd, a few Pt catalysts were proposed to carry out the dehydrogenation of 12H-NEC. Interestingly, Pt/TiO2 was found more active than its Al2O3 counterpart due to a strong metal-supporting interaction that favored electronic transfers from Pt to the support. The rate-determining step was facilitated due to the strengthening of the 4H-NEC-Pt interaction or the weakening of the NEC-Pt adsorption, but no more work was pursued in that direction probably due to the C–N cleavage properties of Pt [364].
Among the Pd-based bimetallic catalysts, Pd2Ru/SiCN was an early example with an improved reactivity compared to the monometallic catalysts [342]. Further developments with a series of Pd–M/TiO2 catalysts (M = Pt, Ru, Ni, Cr, W, Ge) revealed that Pt and Ru were the best co-catalysts. Microwave irradiation was also more efficient than conventional heating at the same temperature due to Pd absorbing the radiation and presenting overheated surfaces [358]. Another series of bimetallic Pd–M (M = Au, Ag, Ru, Rh) supported on rGO revealed the high effectiveness of Au as a co-catalyst (Au > Ru > Rh > Ag). The Au/Pd ratio presented an optimum at Au1Pd1.3, achieving complete conversion and selectivity to NEC in 4 h and reducing by 43% the reaction time comparatively to Pd/rGO. A good catalyst stability was also observed over 5 cycles [369]. Another study on PdAu NP alloys supported on SiO2 showed that Pd3Au was 2.26 times more active than pure Pd and these results were correlated by DFT on (1 1 1) surfaces [361].
Pd alloys with non-noble metals were also studied with bimetallic Pd x Cu y /rGO catalysts. Pd1.2Cu showed 100% selectivity for NEC in 7 h, being as efficient as Pd/rGO while reducing the Pd amount. The catalytic activity was linked to the particle size and the amount of Cu: from 0 to 50% Cu replacement, no variation of the Pd binding energy was measured, indicating similar catalytic activities to pure Pd. Conversely, above 50% Cu addition induced a decrease in catalytic activity due to electronic transfers to Cu [370]. A recent PdNi/KIT-6 catalyst produced by sonochemistry was able to anchor ultrafine NP on the Si–OH surface. Pd4Ni1/KIT-6 catalyst was 1.7 times more active than Pd/KIT-6 at 180 °C due to electron transfers between Pd and Ni and retained more than 90% stability over 5 cycles [371].
Hydrides were also used as supports and dopants for noble metal-free dehydrogenation catalysts. A Co–B–YH3/Al2O3 catalytic system enabled activities comparable to Pd catalysts, but a gradual loss of performance was observed over 5 cycles due to phase detachment from the support [349]. Lastly, a LaNi5.5 catalyst composed of a LaNi5 core and a Ni-rich shell was active for dehydrogenation at 200 °C in 4 h. Due to the low pressure and high temperature, the LaNi5 bonding sites were empty during the dehydrogenation, which promoted the migration and desorption of H2 [350].
-
Conclusion
NEC has been the go-to academic LOHC with the most recent work originating from research groups in Asia, exhibiting great promises as an LOHC with comparative performances to DBT and a reduced enthalpy cost. Nonetheless, selectivity is still an issue due to C–N bond cleavage during the reaction at higher temperatures. In addition, the solid phase hydrogenated compound might complicate the application of this LOHC, although the presence of different isomers can form a liquid eutectic mixture at room temperature. Finally, its high toxicity might impede its further development.
4.2.5.2.2 Perhydro-Phenanzine/Phenazine (12H-PHE/PHE)
Polycyclic aromatic molecules containing 2 N atoms or more included in 6-membered rings have been studied by Cooper up to 2012 [219]. Numerous structures were proposed, in particular phenantrolines, dipyridils, bipyrimidines, quinazoline, terpyridines and naphtyridines (Fig. 18).
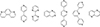 |
Figure 18 Examples of structures studied by Cooper From left to right: 4,7-phenantroline, 4,4-dipyridil, 2,2’-bipyrimidine, quinazoline, 4,2’:5’,4’’-terpyridine, 4,2’:6’,4’’-terpyridine and 1,5-naphthyridine [219]. |
Most studied systems were unreactive or unselective due to incomplete reactions, side-reactions like ring-opening except for the 4,7-Phenantroline for which Pd/C evolved more than 90% of the H2 capacity in 8 h at 250 °C. No follow-up work was found in the literature.
12H-PHE/PHE is a recent LOHC couple that possesses high gravimetric and volumetric densities (7.2 wt.%H2 and 69 gH2/L) (23). It is expensive (26 €/kg) but its synthesis from lignin was demonstrated [342]. Nonetheless, PHE is solid up to 172 °C, requiring dilution in solvents to perform the hydrogenation and dehydrogenation reactions.
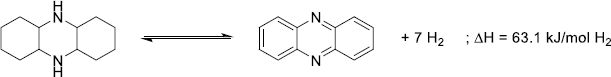 (23)
(23)
Pd2Ru/SiCN completely catalyzed both hydrogenation in solvent conditions (dioxane/water) in 24 h at 115 °C and 50 bar H2 and dehydrogenation in solvent conditions (diglyme) in 24 h at 190 °C, hence solvent separation is needed after the reaction. Further development of the 12H-PHE/PHE system would require solvent-free conditions, but no advances have been reported to this date.
4.2.5.3 O-heterocycles
O-heterocycles are less advantageous than N-heterocycles as the inclusion of an O atom in a cycle does not store hydrogen during the hydrogenation. However, the addition of O atoms modifies the thermodynamics of the aromatic rings in a similar fashion to N atom addition as shown by the comparison of the dehydrogenation enthalpies performed by Cooper using DFT modeling (Fig. 19) [219, 328].
 |
Figure 19 Comparison of the inclusion of heteroatoms on the dehydrogenation enthalpy (kJ/molH2) [219]. |
While the thermodynamics gain is smaller for O-heterocycles than for N-heterocycles, O-heterocycles still have lower dehydrogenation enthalpies than homocyclic LOHC and could prove useful to tune the physico-chemical properties of the LOHC and access bio-based structures.
Dibenzofuran, the analogous structure of NEC, possesses high gravimetric and volumetric densities (6.7 wt.%H2 and 67 gH2/L). Early results presented its hydrogenation with a Ru/LiAl5O8 catalyst, yielding 90% of the hydrogenated carrier at 100 °C and 60 bar H2 and 10% of hydrogenolysed products [219]. Here, the cleavage of the C–O bond by H2 for O-heterocycles required the hydrogenation of the rings prior to the hydrogenolysis (Fig. 20) [372].
 |
Figure 20 Simplified mechanism of the hydrogenation followed by the hydrodeoxygenation of dibenzofuran. |
In addition, numerous articles reported the hydrodeoxygenation of Dibenzofuran with non-noble NiMo/Al2O3 [373] or noble Pd/COK-12 [374] metal catalysts. In particular, Pt, Pd, or Ru/SiO2 catalysts were used to probe the metal activity for hydrogenation and hydrogenolysis. Ru was the most active metal with a high hydrogenolysis capacity while Pt showed a higher selectivity to O-heterocycles in the same conditions but slower hydrogenation activity [375].
On the contrary, the dehydrogenation was performed with a Pd/C catalyst at 225 °C, evolving 60% H2 in 24 h without hydrogenolysis. Moreover, during the dehydrogenation, multiple isomers of the hydrogenated products were formed from a single isomer, indicating an isomerization mechanism similar to NEC. Therefore, hydrogenolysis observed during the hydrogenation could arise from a combination of the Ru catalyst and the high H2 pressure [219]. Thus, except if the hydrogenation is carried out at low pressure with selective catalytic systems, O-heterocycles would show poor cycling performances as LOHC. Recent developments presented the selective hydrogenation (>99%) of eutectic mixtures containing diphenylether with a Pd/Al2O3 catalyst at 50 bar and 120 °C in 189 h and especially an Rh/C catalyst at 20 bar H2 and 60 °C in 18 h [376]. These results could spark new interest in O-heterocycles, but no further development has been published to date.
The Sections 4.2.5.1 to 4.2.5.3 showed the advances of molecules unanimously reported as LOHC in the literature. To further the discussion around new potential LOHC structures, acceptor-less hydrogenation and dehydrogenation reactions with their respective catalytic systems will be presented. Moreover, the discussion will be centered on heterogeneous catalytic systems when possible due to their industrial relevance compared to homogenous systems.
4.2.5.4 Primary alcohol/Aldehydes-esters-carboxylic acids
The irreversible decomposition of alcohols by reforming produces H2, but the reversible hydrogenation/dehydrogenation of primary and secondary alcohols would be more appropriate for the LOHC technology and pave the way for easily bio-sourced LOHC structures [377]. Here, the different heterogeneous catalysts used for the hydrogenation of carbonyl compounds will be reviewed and then specific catalysts for the dehydrogenation of Ethanol (EtOH) will be discussed. EtOH was chosen due to its chemical simplicity that allows studying its dehydrogenation into Acetaldehyde (ACE), Acetic acid (AcOH), and Ethyl Acetate (EtOAc) (24).
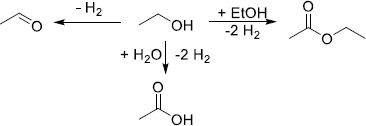 (24)
(24)
In addition, contrary to MeOH which contains only one carbon, EtOH possesses a second carbon in β of the oxygen atom that enables a number of side reactions such as aldolization-crotonisation, intramolecular dehydration, and etherification, making it ideal to study the effect of the catalyst on the reaction.
4.2.5.4.1 Hydrogenation of carbonyl compounds
The classic approach for the hydrogenation of carbonyl compounds to alcohols was the stoichiometric reduction with strong reducing agents like LiAlH4 or boranes [378, 379]. However, the formation of stoichiometric by-products did not comply with the rules of green chemistry, promoting the research of clean hydrogenation procedures. Homogeneous catalysis was able to perform the hydrogenation with a variety of metals using H2 as a reducing agent, but it will not be discussed further. Here, an overview of the active metals and co-catalysts used for the hydrogenation of carbonyl compounds with heterogeneous catalysts will be briefly presented.
An early example of catalytic hydrogenation was published by Sabatier and Sanderens in 1903. Using a Ni catalyst, the hydrogenation of aldehydes and ketones was performed in the gas phase at temperatures as low as 90 °C. Cu was deemed inefficient due to its capacity to catalyze the reverse reaction while Co and Pt were less active than Ni [380]. Since then, heterogeneous catalytic systems based on platinum group metals (Pt, Pd, Ru, Rh, Ir, and Re) are well known to achieve the hydrogenation of aldehydes, esters, or carboxylic acids [381, 382]. Recently, Pt/CeO2–ZrO2 catalyzed the complete hydrogenation of carbonyl compounds at room temperature and 1 bar H2 in 2–4 h. Similarly, noble metal-free catalysts based on Au [383], Cu [384], Ni [385], or Co [386] catalyzed the hydrogenation of carbonyl compounds to the corresponding alcohols. In particular, Cu showed relevant activity for ester hydrogenation, and various co-catalysts and catalysts such as B [387], Ce [384], La [388], Fe [389], In [390], Zn [391], Pd [392], Ag [393], or Au [394] were probed to tune the electron density, catalyst stability and dispersion of Cu as pure Cu catalysts were prone to deactivation by oxidation at high temperature and pressure. Moreover, similar to other LOHCs, small particles, metal-support interaction (for example Pt/TiO2), catalyst wettability and metal reduction state were key to increasing the kinetics [382, 390]. Finally, LOHC-support interactions on basic/acid sites directed the selectivity and high specific surface allowed for the synthesis of smaller active metal NP [395].
4.2.5.4.2 Primary alcohols/aldehydes
Intramolecular dehydrogenation of ethanol yields acetaldehyde following equation (25), for subpar gravimetric and volumetric densities of 4.3 wt.%H2 and 34 gH2/L respectively. In addition, the dehydrogenation enthalpy is similar to that of homocyclic structures due to little stabilization of the dehydrogenated structure [396, 397].
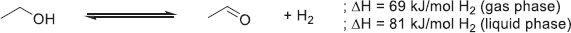 (25)
(25)
An early example of EtOH dehydrogenation was performed in a diluted gas phase by Sabatier and Sanderens with reduced copper at 250 °C, producing pure H2 and a mixture of alcohol and aldehyde in the condensed phase [380]. Further development of Cu catalysts showed a dependence on the catalyst activity due to the copper salt precursor (copper nitrate), the promoter (Co and Cr), and the support material (asbestos). In the optimized conditions (275 °C), 50% EtOH was converted with 85% selectivity to ACE, 9.6% to Ethyl acetate, and Acetic acid was also observed. In addition, the catalyst lost 14% activity after 28 h due to coke deposition, but its regeneration was possible by using H2 [398]. Since then, multiple supports were tested in order to tune the acidic/basic sites and stabilize the active Cu NP. A Cu supported on N-rich carbon achieved 98% selectivity to ACE due to its affinity for well-dispersed Cu NP and its enhanced adsorption properties promoted by the nitrogen atoms [399]. SiO2/SiC and C/SiC supports were then developed as the Si–OH groups promoted the dispersion of the NP [400]. The C/SiC support was advantageous due to its improved desorption properties that blocked side reactions. At 280 °C, the comparison of Cu/SiO2/SiC and Cu/C/SiC showed that the former achieved the best conversion (81% vs. 66% resp.) while the latter was the most selective (94% vs. 99% resp.) [401]. Metal oxides and HT structures were also tested as supports for Cu and revealed that the O–H EtOH bond breaking on support acidic sites was the rate-determining step. However, the rate-determining step was also the Cα–H cleavage depending on the reaction conditions (EtOH pressure, strongly acidic catalysts) [402]. In addition, chemically inert supports like ZnAl2O4 showed high conversion and selectivity (90% and 95% resp.) at 300 °C, but higher temperatures promoted dehydration and coke deposition [403]. Finally, Cu supported on KIT-6 mesoporous silica was modified to obtain moderate acid sites on the surface and to optimally distribute the Cu species, enabling superior EtOH conversion and ACE selectivity (96.8% and >99% resp.) at 250 °C with Cα–H cleavage as the rate-determining step [404].
Other active metals such as Ru and other PGM [405, 406], Ag [407], Au [408], and Co [409] were tested with less success. Selectivity was usually an issue for PGM catalysts due to their higher tendency to promote condensation and dehydration reactions [405, 406]. While the conversion was originally low for Ag/HT (17% in 72 h) [407], recent developments on Ag catalysts supported on SiO2 and CeO2–SiO2 allowed for better dispersion of the Ag NP, increasing at 300 °C the conversion of EtOH to 50% with 95–100% selectivity to ACE in a fixed-bed reactor [410]. Further work revealed the necessary concerted mechanism between Si–OH and Ag sites to activate the Ethanol O–H bond [411]. Conversely, Au/TiO2 followed a different mechanism with the adsorption preference of etoxy species on Ti4+, the promotion of the Cα–H cleavage by the support, and Au NP activation by spillover [408]. Later, Au supported on ZnZrOx catalyzed the selective conversion of EtOH to ACE at 300 °C with a yield of 60%. Nevertheless, higher temperatures showed a decrease in ACE due to Acetone formation [412]. Finally, while early attempts with Co catalysts mainly induced the reforming of the carrier [413], recent advances proposed Co supported on N-doped carbon catalysts, with an EtOH conversion of 66% and a selectivity of 84% to ACE at 400 °C. Nonetheless, the dehydration to ethylene was also observed as a competing reaction attributed to the presence of oxidized Co species formed during the reaction [409].
-
Conclusion
The dehydrogenation of primary alcohols to aldehyde seems rather challenging as dimerization (ester and ether formation), dehydration, and overoxidation are side reactions that are difficult to overcome. From a practical standpoint, dimerization is usually limited by diluting EtOH in a carrier vector and overoxidation could in principle be prevented by ensuring that the reaction medium is air- and water-free. Nonetheless, dehydration would still occur due to the high reaction temperature and promotion by acid sites on the support.
4.2.5.4.3 Primary alcohols/Carboxylic acids
Acetic acid is a potential LOHC that can be obtained by the dehydrogenation of equimolar ethanol–water solutions, reaching densities of 6.3 wt.%H2 and 54 gH2/L (26) with lower dehydrogenation enthalpies than homocyclic and heterocyclic systems [397, 414]:
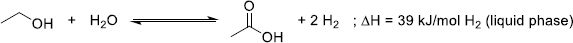 (26)
(26)
Originally, the conversion of primary alcohol to a carboxylic acid required carrier oxidation, using either air or harsher chemicals as oxidant and Au-based heterogeneous catalysts in the liquid phase [415]. However, the presence of O2 can also displace the reaction equilibrium by unfavorably converting H2 to H2O by combustion reaction (1). The first acceptorless dehydrogenation was carried out by Balaraman et al. with a homogeneous Ru catalyst in the presence of a NaOH aqueous solution where water acted as the oxygen donor, producing carboxylate salts with the concomitant release of H2 [416]. This reactivity was based on Cannizarro or Tishchenko reactions that allowed for the disproportionation of aldehydes in alkaline conditions by direct hydride transfer or ester formation and hydrolysis respectively to form the primary alcohols and carboxylate salts (27) [417, 418]. Hence, any catalyst converting the selective dehydrogenation of primary alcohols to aldehydes could in principle be relevant for this reaction.
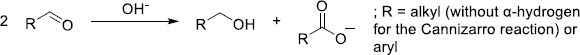 (27)
(27)
The first example of heterogeneous acceptor-less dehydrogenation used Rh/C in a closed vessel with 2.2 equivalents of base in H2O at 100 °C under Ar, obtaining 100% conversion but limited selectivity (<55%) to the carboxylic acid with NaOH in 24 h [419]. Subsequent work by the same group assessed the noble metal efficiency for the reaction, with the activity order Pd > Rh > Pt > Ru/C. The effect of the base was also investigated with the reactivity order NaOH > KOH > LiOH > Na2CO3 > NaHCO3 > no base. In addition, the reaction was performed under reduced pressure (0.8 bar) and 80 °C to facilitate the removal of H2. Reactions on both aliphatics and benzylic alcohols achieved quantitative yields in 6 h [420]. Further catalytic development on noble metals was scarce. Recent advances reported Pd/NiO NP able to dehydrogenate benzylic alcohols converted with up to 97% yields in the presence of 1.1 equivalents of KOH in 6 h at 110 °C. 50–70% yields could also be achieved for aliphatic alcohols with longer reaction times. Moreover, the catalyst showed only little activity loss (<10%) over 6 cycles [421]. Finally, supported Ru homogeneous catalysts also catalyzed the reaction with 1.1 equivalents of KOH at 110 °C, achieving up to 99% conversion and selectivity for both benzyl alcohols and aliphatic alcohols. The catalyst was stable for 20 cycles, retaining 99% of its original activity [422].
Noble metal-free catalysts were also developed. ZnO with two equivalents of KOH catalyzed the conversion at 164 °C in 36 h in 65–85% yields for aliphatic product and similar yields were obtained for benzylic alcohols in 18 h. The reaction pathway occurred through a zinc alkoxide species that further dehydrogenated to aldehydes and esters through a Tishchenko mechanism [423]. Co catalysts were also used for the clean conversion of benzylic alcohol to the corresponding carboxylic acids. Co/N-doped CNT fully converted benzyl alcohol with 99% selectivity to the benzoic acid in 24 h at 100 °C. The conversion of aliphatic alcohols was generally slower but good yields (>80%) were obtained if the base amount and time were increased [424]. Similarly, Co/N-doped carbon achieved 100% conversion of Benzyl alcohol and 88% selectivity to benzoic acid when reacted with 1.2 equivalents of KOH in 24 h at 164 °C. The aliphatic alcohols reactivity was lower, but 80% yields were achieved in 36 h. In addition, the catalyst was stable for 15 cycles without a significant loss of activity and was easily retrieved from the reaction mixture by magnetic separation [425]. Recently, a bimetallic ZnCoO x rod-like catalyst with 1.2 eq NaOH achieved up to 96% yield for benzylic alcohols in 18 h at 135 °C. Aliphatic alcohols were only slightly less reactive, with 85–88% yields in 20 h. Gram-scale (5 g) reactions were performed for both benzyl and aliphatic substrates in similar yields. Finally, the catalyst showed excellent stability over 10 cycles. Interestingly, the reaction pathway was here more akin to a Cannizarro-type reaction as no ester formation and disproportionation of the aldehyde to the alcohol and the acid was observed [426].
-
Conclusion
The acceptor-less heterogeneous dehydrogenation of primary alcohols to carboxylic acids has only recently been studied. Nonetheless, some system limitations must be overcome in order to use this reaction in a LOHC system. Indeed, the use of stoichiometric amount of base, the reaction in solvents as well and slow kinetics for aliphatic alcohols are bottlenecks that still need to be addressed.
4.2.5.4.4 Primary alcohols/Esters
Esterification of primary alcohols can be afforded by intermolecular reaction with homocoupling (28)-A and heterocoupling (28)-B variations or intramolecular reaction (28)-C.
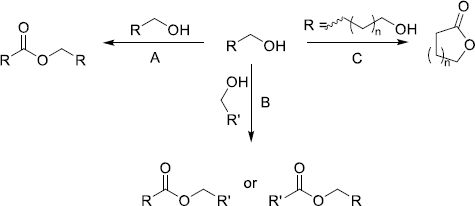 (28)
(28)
Here only acceptorless catalytic systems developed for dehydrogenation will be discussed. Variations B and C will be studied through the dehydrogenation of the ethanediol+ethanol/diethyloxalate and 1,4-butanediol/γ-butyrolactone couples respectively.
Intermolecular homo-esters: The couple ethanol/ethyl acetate (EtOH/AcOEt) has similar gravimetric and volumetric densities to the EtOH/ACE couple (4.3 wt.%H2 and 34 gH2/L resp.). However, its dehydrogenation enthalpy is much lower than the latter due to the condensation of ACE with EtOH being exothermic overall (29) [397, 427].
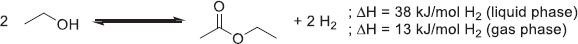 (29)
(29)
In 1903, Sabatier and Sanderens observed the conversion of EtOH to ACE with a tenth of EtOAc as a by-product [380]. The full dehydrogenation of primary alcohol to ester was achieved in 1964 by Franckaerts and Froment using CuO–CoO–Cr2O3/asbestos. Kinetics showed that the rate-determining step was the adsorption of two EtOH molecules on adjacent sites prior to the reaction [428]. Since then, Cu/ZrO2 and Cu/ZnO catalysts reached respectively 27% and 28% selectivity at 50% EtOH conversion with the formation of intermediates like ACE and side-products such as diethylether, acetone, and other C4-products. Support modification with KOH suppressed the formation of diethylether on acidic alumina. In addition, the selectivity was dependent on the catalyst Cu loading, and investigation of the catalytic site showed the importance of the Cu reduction state and the support (pristine ZrO2). Cu was necessary for the dehydrogenation of EtOH to ACE while pure ZrO2 catalyzed the dehydrogenation of ACE to EtOAc, achieving 100% selectivity at 160 °C [429]. These findings were confirmed by further studies that highlighted the primordial role of Cu+ species (i.e. unsaturated sites on the Cu NP like edges or steps as well as Cu atoms bonded to the metal oxide) to enhance the reactivity [430].
The role of the support was also investigated by various studies that revealed that basic substrates with an acidic metal center and O2− basic sites like ZrO2, CeO2, HT, or MgO were much more active and selective than acidic substrates like TiO2, SiO2, and Al2O3 [431, 432]. Mixing multiple metal oxides to tune the support properties drastically increased the catalyst dehydrogenation properties. In the case of the addition of ZnO and ZrO2 with Al2O3, the conversion increased to 66% and the selectivity to 85% at 220 °C [433]. Key steps of the mechanism was also elucidated on this catalyst, rationalizing the multi-step mechanism and the by-products formation pathways. Finally, dehydrogenation under different pressures revealed a positive effect on selectivity, indicating that by-product formation was dependent on the accumulation of ACE. Indeed, when increasing the pressure to 8 bar at 200 °C, up to 93% selectivity was achieved, but the conversion was reduced by 9% [434].
Cu–Cr catalysts also showed high conversion and selectivity and at 200 °C, Cu/Cr2O3 catalyzed the formation of EtOAc with 95% selectivity, highlighting the role of Brønsted acid sites on the selectivity [435]. Further catalytic development introduced the doping of alumina with BaCrO4 to form a CuCrO4/CuO/Cu/BaCrO4/Al2O3 catalyst that increased the EtOH conversion to 65–70% and the selectivity above 98% at 220 °C and 20 bar in flow conditions. Here, the rate-determining step for each released H2 molecule was assessed, revealing that the first rate-determining step was the dissociative adsorption of EtOH to produce an adsorbed ethoxy group and the second rate-determining step was the condensation of ACE with EtOH in a hemiacetal that acted as an intermediate before further dehydrogenation to EtOAc (30) [436].
 (30)
(30)
The rate-determining step was catalyst-dependent as a DFT study on Cu (1 1 1) reported that the rate-determining step was the dissociation of EtOH [437], while the condensation of an alcohol and an aldehyde was reported for Cu/ZrO2 [432]. In addition, each component of the catalytic system has a dedicated role that could be summarized as follow: reduced Cu0 for the dehydrogenation activity of EtOH, Cu+-metal oxide interface as the preferential site for EtOH adsorption and the support for the catalysis of the O-H bond cleavage to form Cu-alkoxides and side-reactions like dehydration [432, 438]. In particular, tuning the Cu0/Cu+ ratio by the modification of the NP size was paramount for the selectivity as ACE would form at low ratios while EtOAc would form at higher ratios [439]. In fact, the effect of size modification (i.e. Cu0/Cu+ ratio modification) was also promoted by support replacement [440], support phase modification [441] or Cu content adjustment [429].
As other metals such as Au, Pd, Pt, Co, or Ag and their alloys were active for the dehydrogenation of EtOH to ACE (see para “Primary alcohols/aldehydes” from Sect. 4.2.5.4), these catalysts could be in principle active for the dehydrogenation of EtOH to EtOAc, but only limited examples have been reported. The comparison of active metals supported on ZrO2 for the dehydrogenation of EtOH to AcOEt was first performed with a reactivity order Cu > Ni > Ag > Pt > Ru > Ir > Pd ≈ without metal [432]. Interestingly, this order was dependent on the support as a series of catalysts supported on SnO2 had a reversed order of reactivity with a surprisingly inactive Cu catalyst: Pt > Rh > Ir > Pd > Re > Ru > Ag = Ni = Co = Cu = 0. Here, the synergistic effect of basic supports with Pt was attributed to the activation of the aldehyde species by the acidic Sn4+ metal center and O2- basic sites. Other supports were tested with their activity ranked in the order SnO2 > ZrO2 > CeO2 > Nb2O5 > TiO2 > C = Al2O3 = SiO2 = HBEA zeolite = MgO = 0 [442]. Finally, Pd/ZnO was also able to achieve the dehydrogenation to EtOAc, but the reaction was not selective as ACE was the main by-product, potentially due to the Pd structure that increased the stability of ACE on the surface [443].
-
Conclusion
Homo-esterification presents numerous advantages like low-cost Cu-based catalysts and a high selectivity tunability by modifying the support, and the distribution of tge Cu species as well as adding promoters. However, due to the multistep nature of the mechanism, different stable intermediates can form such as aldehydes, which can limit the control of the system. Finally, dehydration at high temperatures in the presence of acidic species is still a major side reaction.
Intermolecular hetero-esters: Dehydrogenative cross-esterification is challenging and still in its infancy. Indeed, while process techniques such as drop-wise addition facilitated the cross-coupling of a primary alcohol with a secondary or tertiary alcohol, homocoupling was difficult to overcome when mixing two different primary alcohols. Attempts for direct cross-esterification were also performed with heterogeneous transition metal sulfides MoS2 and WS2 at 230 °C, achieving 52% conversion in 24 h under 5 bar He pressure but a mixture of symmetrical and asymmetrical esters was obtained [444]. The most successful examples of heterocoupling were performed with homogeneous catalysts, using P and N bidentate or tridentate pincer ligands as well as monodentate N-Heterocyclic Carbene (NHC) ligands to control the coordination sphere of the molecules on the catalyst. Examples of such ligands are presented in Figure 21.
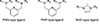 |
Figure 21 Classic ligands used in homogeneous catalytic systems for the acceptorless dehydrogenation reaction. R1, R2, R3, R4 = H, alkyls, aromatics. |
 |
Figure 22 O and N mixed moieties obtained by the coupling of alcohols and amines |
The first example to yield asymmetrical esters relied on the trans-esterification of symmetric esters using a secondary alcohol with Ru PNN-pincer catalyst in 17–36 h at 135 °C with up to 95% selectivity [445]. Direct cross-coupling of primary alcohols was originally performed with a dimeric Rh homogeneous catalyst in the presence of 0.5 equivalent of NaHCO3, achieving 67–97% yields on a variety of aromatic substrates [446]. Recent advances suggested an Mn PNN-pincer catalyst in the presence of a base to yield the cross-coupling of primary alcohols in 71–95% yields and trans-esterification with high selectivity (70%) [447]. To date, the best example was published by Zhou et al. with the ethylene glycol+ethanol/diethyloxalate couple using a Ru PNP-pincer complex under solvent-free and base-free conditions to yield a reversible mixture of different esters including symmetrical esters like EtOAc (13%) and other asymmetrical esters like ethane-1,2-diyl diacetate (18.5%) (31) [448].
 (31)
(31)
While such structures present great interest in tuning the hydrogen capacity of the LOHC systems, progress on that account has been limited due to a lack of selective heterogeneous dehydrogenation catalysts for asymmetrical esterification.
Intramolecular esters/Lactones: Intramolecular esterification has been mainly studied through the 1,4-butanediol/γ-butyrolactone (BDO/GBL) couple as succinic acid shows great promise as a platform chemical derived from biomass [449]. In addition, BDO can store up to 4.4 wt.%H2 and 45 gH2/L and presents an attractive dehydrogenation enthalpy [397, 450–452]. Due to its high boiling point (230 °C), BDO dehydrogenation was studied in both liquid- and gas phases with most literature focusing on the gas phase. The dehydrogenation for both phases will be reviewed hereinafter.
 (32)
(32)
-
Gas-phase
Early patents for the dehydrogenation were submitted from 1990 to 2000 with catalysts based on the Cu–Zn [453], Cu/Cr/Mn/Ba/Na [454], Cu/SiO2–CaO [455] and Bi-doped Pt [456] systems which reported conversion and selectivity above 98%. A later comparative study revealed that metal activity on ZrO2 was following the order Cu > Ni > Ag > Pt > Ru > Ir > Pd = no metal [432].
A series of CuO:ZnO:ZrO2:Al2O3 catalysts was originally developed to assess the impact of each constituent on the conversion and selectivity of the dehydrogenation. A hemiacetal intermediate was proposed to be formed during the dehydrogenation, similar to the EtOAc mechanism with Cu0 as the active phase. THF formation was attributed to acidic sites on Al2O3 that could be suppressed by ZnO doping. In the optimized conditions, a (6:1:2:2) CuO:ZnO:ZrO2:Al2O3 achieved up to 84% conversion and 98% selectivity [457]. Kinetics experiments showed that the alcohol–aldehyde condensation to form a hemiacetal was the rate-determining step of the system (33) [432].
 (33)
(33)
The influence of acidic and basic sites of the support was predominantly affecting the selectivity of the reaction. Especially, the dehydration of the intermediate hemiacetal to THF due to strong acidic sites of the support and the concerted effect of the temperature was also reported by numerous works [458–460]. However, both acidic and basic sites were required in similar proportions to dissociate the O–H bond in a tandem mechanism in order to form Cu–alkoxide species. With Cu as an active metal, the support reactivity and selectivity order were ZrO2 > CeO2 > Al2O3 > SiO2 > TiO2, exemplifying the beneficial effect of basic supports like ZrO2 that possess relative spatial proximity of their acidic metallic sites (Zr4+) and their basic sites (O2−) [432]. Similarly, CeO2 and MgO achieved >99% conversion and >99% selectivity [458, 460–462]. In addition, akin to ester dehydrogenation, lactone formation required a fine-tuning of the Cu+/Cu0 ratio with dopants like La, Ba, Ca, Sr, or Mg that electronically enriched the Cu+ sites in contact with the support, facilitated the reduction of Cu and prevented the sintering of the NP. The addition of La as a promoter increased the yield of GBL from 51 to 93% and allowed for a stable catalytic activity for 15 h on stream while Ba addition on a Cu/SiO2 catalyst achieved 100% conversion and 99.6% selectivity at 240 °C [459, 463]. The effect of the particle size on the Cu+/Cu0 ratio was also visible on a series of Cu/CeO2 catalysts with Cu content ranging from 5 to 20%. At high Cu content, the bigger Cu particles were less active, but the selectivity for GBL increased to >99% [460, 464]. In addition, continuous reduction of Cu by H2 was observed on Cu/SiO2, rationalizing its excellent catalytic performance stability (95.8% conversion and 99% respectively) over 400 h on stream [464]. While SiO2 was less selective than basic oxides, mesoporous SBA-15 presented weak acid sites that were beneficial for the tandem O–H bond cleavage but not potent enough to induce major dehydration, reaching 100% conversion and 98% selectivity [465].
-
Liquid-phase
Liquid-phase dehydrogenation was much less studied than the gas-phase dehydrogenation of BDO to GBL, often due to its lower kinetics: for the (6:1:2:2) CuO:ZnO:ZrO2:Al2O3, the dehydrogenation at 200 °C had a similar selectivity but the full conversion was achieved for a residence time 6 to 7 times longer [457]. Interestingly, the Cu activity in the liquid-phase was much lower than that of noble metals and an early study showed that GBL was obtained in 94% yield with Ru/AlO(OH) at 110 °C in 32 h and in dilute conditions [466]. A comparative study of metal activity on SnO2 at 180 °C highlighted that the reactivity order of was Pt > Ir > Ni > Pd > Co > Rh > Cu > Re > Ag while it was Pt > Rh > Pd when supported on rutile TiO2 in photocatalytic conditions [467, 468]. Interestingly, Pt required different supports than Cu: ZrO2 or MgO were inactive while SnO2 or Al2O3 catalyzed the reaction. In solvent-free conditions, Pt/SnO2 achieved 100% conversion and 80% selectivity in 36 h at 180 °C. The exceptional activity of this catalyst was linked to Sn4+ acid sites that activated the aldehyde intermediate [467]. Photocatalysis at room temperature in 1 h with different TiO2 phases showed the lactone instability on the anatase phase while the rutile phase cleanly catalyzed the dehydrogenation. By adding Al2O3 with weak acid sites, the photocatalytic mechanism was promoted while strong acid sites protonated titanate nanotube addition was linked to dehydration and coupling product formation. Unfortunately, the dehydrogenation was much more active for the aromatic phtalides than BDO achieving 90% and 20% conversion respectively [468].
-
Conclusion
Lactonization was often reported in the literature using heterogeneous catalytic systems based on Cu and noble metals. Gas-phase dehydrogenation (> 200 °C) presents higher kinetics than liquid-phase dehydrogenation due to the higher reaction temperature, but some limiting diffusion phenomenon might also take place in the liquid-phase, hindering the reaction. Moreover, as thermodynamics favors the formation of 5-membered rings, the BDO/GBL couple might enable the research of low dehydrogenation enthalpy LOHC.
4.2.5.4.5 Secondary alcohols/Ketones
Ketones and secondary alcohols are abundant in nature and are hence of great interest in order to produce LOHC from renewable feedstock [469]. In addition, Isopropanol (IPA) and acetone industrial productions total more than 8 million tons, showing their availability [470]. Nevertheless, IPA has moderate gravimetric and volumetric densities (3.3 wt.%H2 and 26 gH2/L) as well as a dehydrogenation enthalpy equivalent to homocyclic systems (34) [397, 471]. Moreover, contrary to primary alcohols, secondary alcohols are less sensitive to overoxidation, facilitating their storage and recyclability. However, they can be extremely reactive with peroxides and are prone to aldolization in alkaline conditions.
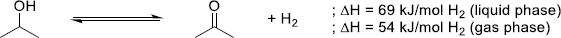 (34)
(34)
The earliest example of IPA dehydrogenation to acetone was reported by Sabatier and Sanderens with a Cu heterogeneous catalyst that achieved up to 75% acetone yield at 420 °C [380]. During the 20th century, the dehydrogenation of IPA to acetone was often performed in a reactive distillation setup with Ni Raney at 82.5 °C, using the boiling point difference of Acetone (56 °C) and IPA (82.4 °C) to remove acetone from the reaction mixture in order to displace the reaction equilibrium and increase the reaction rate [472–474]. This setup allowed for the design of a chemical heat pump able to store waste heat by using H2 as an energy vector. Nevertheless, the conversion was quite slow with less than 5% yield after 6 h at 80 °C [474]. A comparison at 90 and 100 °C revealed that the reaction proceeded 5–10 times faster in the gas phase and that the reaction rate was less dependent on the acetone concentration [475]. Ru and Ru–Pt supported on activated carbon were also tested and showed an improved conversion from 5 to more than 85% in 2 h at 100 °C due to a better thermodynamic equilibrium displacement when performing the reaction in the liquid film state [476]. A later study reported an improved setup able to evolve up to 5 LH2/h with a Ni Raney catalyst [477].
Gas-phase dehydrogenation was pursued when it became clear that reactive distillation would not yield sufficient activities. Pt, Cu, and their alloy supported on activated carbon were found to be reactive in the order Pt > Pt–Cu > Cu at 175 °C. As expected, kinetics experiments showed that the reaction rate was favored by IPA concentration and slightly disfavored by H2 and acetone concentrations. Moreover, the rate-determining step was attributed to the cleavage of the hydroxyl bond [478, 479]. Cu-based catalysts were principally studied due to their activity above 200 °C. The microstructure of the carbon had no influence on the dehydrogenation rate but modified the selectivity, with platelets achieving 100% selectivity to acetone at 200 °C. Ce addition increased the activity by 6 but favored the dehydration reaction to propene [480]. Later, Cu+ was described as the active species and the addition of NiO increased the charge transfers between Cu+ and Ni3+, further improving the basicity of the catalyst which allowed for a better acetone desorption [481]. Recent catalyst composition included CuO/TiO2–ZrO2 with PtO as a promoter to increase the basicity and reactivity of the support [482] while a CuO supported on a carbonized MOF achieved 100% conversion and selectivity in a fixed bed reactor at 275 °C by tuning the flux rate [483].
-
Conclusion
Secondary alcohols are intrinsically limited by their low H2 densities if no H2 can be exploited on the β-carbons. However, the case of IPA/acetone is highly interesting as it shows that the dehydrogenation of secondary alcohols can happen with noble metal-free heterogeneous catalysts at the operating temperature of PEM-FC, which would allow for excellent system integration. Unfortunately, the kinetics are still quite low, and most achieved work has been related to system development. Therefore, catalytic development of such reactivity would be of great interest.
4.2.5.5 Alcohol and amine couplings
Alcohol and amine couplings can produce various chemical moieties such as amides, ureas, imides, or carbamides which enable the innovative design of alternative structures for the LOHC technology. In addition, amino acids could be platform chemicals for the production of renewable bio-based O, N-LOHC.
While no heterogeneous catalyst was reported to date, amides, carbamides, and imides have already been successfully reversibly produced by the dehydrogenation/hydrogenation of alcohols and amines in homogeneous conditions. These reactions will be presented to broaden the perspectives for future LOHC systems.
4.2.5.5.1 Amides
Amide synthesis applications range from polymer synthesis [484] to bio-engineering of polypeptides [485]. The first catalyst to achieve the dehydrogenative coupling of a primary amine and a primary alcohol to an amide was a Ru PNN-pincer (Fig. 21) with up to 99% yield in 8 h at 110 °C in toluene for both aromatic and aliphatic compounds. No conversion was observed for the reaction of esters to amides by amine addition [486]. Further articles developed Ru-NHC [484, 487, 488], Ru-PNN [485, 489, 490], and Ru-PNP [491] pincer-type catalysts to achieve better conversions and selectivity on a wide variety of substrates. Most Ru-pincer systems necessitated a basic additive, often KOtBu or NaH, and the amides were obtained in yields ranging from 70 to 98% in 24 h at 110 °C in dilute conditions [484, 488]. Later results reported a Ru PNP-pincer catalyst able to hydrogenate and dehydrogenate in solvent-free conditions with 76% and 60% yields respectively [491] and further development reduced the temperature of dehydrogenation to 35–55 °C [489]. An experimental study coupled with DFT modeling revealed that the amide formation required the stabilization of the catalyst-aldehyde adduct formed during the dehydrogenation of the alcohol so that the nucleophilic attack of the aldehyde by the amine resulted in an intermediate hemiaminal species analogous to the hemiacetal formed during the dehydrogenation of alcohols to esters. Different effects were also probed such as the catalyst loading, the nature and loading of the base, the limitations due to the steric hindrance of the substrate, and the ring size dependence for intramolecular amide formation [492]. Amide intramolecular formation was often studied as an analog of the BDO/GBL couple, but only moderate yields (45–68%) were obtained [484, 488]. To date, the most efficient LOHC systems were based on β-amino alcohols that formed either linear peptides or cyclic dipeptides depending on the bulkiness of the substituents linked to the β-carbon. High bulkiness promoted cyclic dipeptide formation up to 99% yields in 19 h at 135 °C in dioxane [485]. Neat dehydrogenation produced the polyamide in limited yields (48%) while diluted conditions favored the production of the cyclic dimer with selectivity up to 70%. Ethanolamine (6.5 wt.%H2 and 67 gH2/L) was formed back by the hydrogenation of the dehydrogenated reaction mixture in quantitative yields and the presence of polyamides was responsible for an increased reaction time. The stability of the system was limited as a decrease of 25% in conversion for both steps was observed over 3 cycles (35) [490].
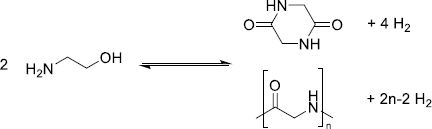 (35)
(35)
Recent developments promoted the use of Mn PNN [493] and PNP [494] pincer catalysts that catalyzed the reaction of symmetric esters with amines to amides in a similar fashion to transesterification [493]. In addition, dimethylethylenediamine and methanol were used as a hydrogenated feedstock to produce the corresponding diamide (36).
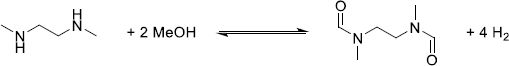 (36)
(36)
The catalytic system was efficient for both hydrogenation and dehydrogenation with complete conversion of the starting material and 86% selectivity [494].
4.2.5.5.2 Carbamides
Carbamide groups are found in numerous natural chemicals such as urea. Their synthesis was achieved by the acceptorless dehydrogenation of ethylendiamine and methanol with Ru PNP [491] and PNN [495] pincer catalysts to form 2-Imidazolidinone (6.5 wt.%H2 and 58 gH2/L using the density of ethylendiamine) (37).
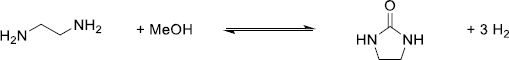 (37)
(37)
The dehydrogenation was performed in 24–48 h with a Ru PNN-pincer catalyst to produce the carbamide, acyclic monoamide, and acyclic diamide species. All obtained products were regenerated in 1.5 days at 170 °C and 60 bar H2 to their hydrogenated constituents with up to 83% yield for ethylenediamine while methanol was always obtained in lower yields [495].
4.2.5.5.3 Imides
Imides and especially aromatic imides are important synthesis intermediates for the fabrication of pigments and are highly valued in high-added value niche applications such as electronics parts [496]. While an early attempt at the hydrogenation of cyclic imides by Cooper was proven unsuccessful using a heterogeneous Ru/LiAl5O8 catalyst at 170 °C [219], recent advances reported the production of cyclic imides through the dehydrogenation of BDO with amines by Mn [497] and Ru [498] PNN pincer catalysts. Early results with a Mn PNN-pincer catalyst and KH as an additive performed the dehydrogenation of BDO with a variety of amines in 60–99% yields in 40 h at 110 °C. In particular, diamines were particularly reactive and produced dicyclic imide structures. Moreover, the high reactivity of GBL with amines was revealed and the mechanism for the coupling could follow simultaneously two reactive pathways, either a direct nucleophilic attack akin to an amide formation from an alcohol and an amine or a ring-opening mechanism due to a nucleophilic attack on GBL. The imide formation then proceeded through a hemiaminal intermediate [497].
Further development with a Ru PNN-pincer catalyst and KOtBu achieved in 40 h at 135 °C and 40 bar H2 the hydrogenation of cyclic imides to BDO and the corresponding aromatics or aliphatic amines in 99% conversion with 99% yields for both BDO and the amines. BDO and ethylendiamine were specially studied due to their high combined gravimetric and volumetric density of 6.7 wt.%H2 and 68 gH2/L (with regard to the density of BDO) (38).
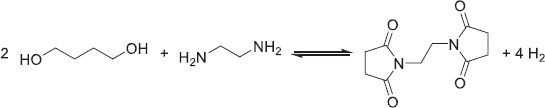 (38)
(38)
The reverse dehydrogenation with the same catalyst in the presence of KOtBu in dioxane achieved 99% conversion and 70% selectivity to the imide, 12% to the lactone, and the rest to oligoamides in 24 h at 120 °C. No complete cycling was performed, but the yield of the cyclic imide was 64% after the second dehydrogenation [498].
-
Conclusion
Alcohol and amine couplings were recently discovered and developed by the Milstein group, opening a key segment of bio-based molecules to the LOHC technology. The main limitations of such systems come from the homogeneous catalysts that require strong bases as additives, the presence of solvents, and the formation of polymeric by-products. Moreover, it should be noted that the extremely low temperature achieved in these studies could be due to the dilution of H2 in an unreactive gas vector, effectively performing the dehydrogenation against a vacuum due to the almost null H2 partial pressure. Therefore, evaluation of these reactivities under 1 bar H2 might strongly modify the required reaction temperature. In addition, H2 dilution in gas might also require gas separation in order to achieve high energy in integrated systems.
4.2.5.6 Amines/Nitriles
H2 storage in the primary amine/nitrile function was proposed by Cooper [219] and later theorized [499] as a hydrogen storage system able to store up to 13.3 wt.%H2, effectively doubling the best gravimetric densities of classic homocyclic LOHC. In particular, LOHC couples like 1,3-propanediamine/malononitrile could reach exceptionally high gravimetric and volumetric densities (10.8 wt.%H2 and 95 gH2/L resp.) at the cost of an increased reaction enthalpy and toxicity (39) [263].
 (39)
(39)
Patents related to the dehydrogenation of amines to imines and nitriles were published in 2010 [500] and 2014 [501] respectively and a start-up named ASEMBLON Inc. was created by the main inventor, E. Naeemi. The start-up developed stationary energy storage systems able to release 99.99% pure H2 in a dual-bladder fuel tank [502].
-
Hydrogenation
The first example of nitrile hydrogenation was published by Sabatier in 1905 with a reduced Ni catalyst that performed the unselective reduction of nitriles with H2 to primary, secondary, and tertiary amines [503]. While numerous catalytic systems have been developed such as colloidal Pd [504], Ni Raney [505], Pt oxide [506], Raney Co [507], Cu2Cr2O5 [508], supported platinum group metals metals [509], metal borides [510], metal alloys [511], the less active Raney nickel and copper chromite are still used industrially due to their lower price. However, their problematic toxicity promoted the development of non-noble transition metals Ni [512], Co [513], Cu [514], and Fe [515] heterogeneous catalysts. Interestingly, most recent systems carried out the hydrogenation in the presence of ammonia in order to thermodynamically disfavor the transamination reaction that produced the dialkyl and trialkylamines compounds. In particular, Ni/SiO2 achieved the complete conversion and 84% selectivity to the monoalkylamine in 5 h at 100 °C and 13 bar H2 in ethanol [516]. Further development highlighted the efficiency of K-NiCo/Al2O3 to selectively (>99.9%) convert Isophthalonitrile to m-Xylylenediamine at 80 °C and 60 bar H2 in the presence of NaOH and in a mixture of Toluene and Methanol [517]. More heterogeneous and homogeneous catalytic systems used to perform the hydrogenation of nitriles to primary amines can be found in the following reviews [518, 519].
-
Dehydrogenation
An early example of amine dehydrogenation was performed with a Mo catalyst that showed amine dehydrogenation with disproportionation, achieving nitrile selectivity inferior to 5% due to imine couplings [520]. Since then, no selective heterogeneous catalysts have been reported for this reaction in solvent-free conditions. Further work relied on Ru [521] and Ir [522] pincer catalysts in order to control the active center. Ir homogeneous catalysts showed great promise to catalyze the reaction when in the presence of a base as a co-catalyst. Depending on the base amount, 97% conversion and 98% selectivity to nitrile at 160 °C in 24 h were obtained, diminishing to 2% nitrile without base [522]. However, due to its high price, Ru was often proposed as an alternative catalyst. The first selective Ru catalyst yielded nitriles in 20–80% rates without oxidant or H2-acceptor in toluene at 110 °C in 24 h [521]. The mechanistic analysis of the Ru NNN-pincer catalyst revealed that a fast dehydrogenation of the imine intermediate was required in order to avoid a nucleophilic addition on the aldimine center resulting in transamination, thus dilution in a solvent facilitated the selectivity to the nitrile coumpounds [523]. These results were confirmed by DFT analysis and showed the impact of steric hindrance of the ligand and the primordial role of the non-covalent interactions such as H-bonding on the thermodynamic stability of the intermediate species [524]. The role of the ligand as a hydride transfer reaction center was exemplified with a Ru-Hexamethylenetetramine (HMTA) catalyst that achieved 90% conversion of aromatic primary amines to the nitriles species in 24 h at 110 °C in toluene [525]. Recent developments achieved similar performances on comparable systems, showing the ligand diversity able to promote this reaction: pyrazole NNN [523], HMTA [526, 527], and NHC-N-P [528]. Lastly, a Ru heterogeneous catalyst supported on the UiO66(Ce) MOF structure achieved 25–90% yields to nitriles in H2O in 16 h at 130 °C. Although the structure seemed stable for 4 cycles, the yield dropped by 50% after 6 cycles due to structural collapse [529].
Electroxidation has also been employed to electro-oxidize amines to nitriles at the anode and produce H2 at the cathode. In 1982, an early example reported a Ni(OH)2 anode in KOH/H2O that converted benzylamine into benzonitrile with a 90% yield in 4 h at 40 °C [530]. Recent examples used NiSe [531] and Ni2Si [532] anodes in KOH/H2O solution at room temperature that achieved the conversion in 3 h with a faradic efficiency over 95% and 40 min with a faradic efficiency over 99% respectively.
-
Conclusion
Primary amines have the highest theoretical gravimetric and volumetric densities of any LOHC, therefore their study seems rather compelling. Nonetheless, these systems are usually limited by their low selectivity due to the transamination reaction. As this side-reaction is particularly prevalent in neat conditions, the development of efficient amine/nitrile based LOHC is limited if no selective catalyst is designed. Moreover, their high toxicity and corrosivity are another barrier to their implementation.
4.2.5.7 S-containing LOHC
S-containing LOHC has been often dismissed due to their high tendency to undergo hydrodesulphurization in the presence of H2 and to strongly inhibit the noble metal catalyst activity by irreversible binding [533]. Early hydrogenation attempts of toluene and naphthalene in the presence of dibenzylthiophene with Pt–Pd/SiO2–Al2O3 revealed the conversion of dibenzylthiophene, but no analysis was performed on the obtained products [534]. Later, hydrogenation with Ru/C yielded no conversion of dibenzothiophene and no study has reported it ever since [372].
A patent filled in 2007 by Naeemi reported the cyclizing dehydrogenation of 1-pentanethiol (5.8 wt.%H2 and 48 gH2/L) to 2-methylthiophene with an Au catalyst (40) [535].
 (40)
(40)
Two-methylthiophene hydrogenation to tetrahydromethylthiophene was later reported with noble metal catalysts exhibiting a better selectivity to the desired product than bimetallic, phosphide, and sulfide catalysts. The selectivity was inversely dependent on the number of basic active sites that favored hydrodesulfurisation, however, no catalyst could efficiently carry out the reaction [536]. No further development was reported to this date.
Recently, the thioesters hydrogenation was achieved with a Ru-acridine homogeneous catalyst with conversion >99% and yields >90% for both the alcohol and the thiol on a wide variety of examples in 36 h at 135 °C and 20 bar H2 in dioxane. Equivalent performances were obtained at 40 bar H2 for the conversion of thiocarbamates and thioamides to the corresponding thiols and the respective amides and amines (41) [537]. However, the comparison of the densities of the thioester analog of EtOAc shows a lowering of both densities due to the atomic weight of the S atom (S-ethyl thioacetate: 3.7 wt.%H2 and 29–48 gH2/L depending on ethanol and thioethyl densities respectively):
 (41)
(41)
The dehydrogenation of alcohols and thiols to thioesters with a similar Ru homogeneous catalyst was reported soon after [538, 539]. Alcohols were converted to esters, while a mixture of alcohol or aldehyde with a thiol yielded mainly the thioester. A strong pressure dependence was observed as the yields dropped from 93% to less than 1% when the H2 pressure was increased from 0 to 1.9 bar.
Interestingly, DFT modeling showed that the kinetic competition between the ester and thiol formation was controlled by the stronger and quasi-irreversible Ru-thiolate bonding due to the stronger acidity of the thiol compared to the alcohol. Although the ester was the most stable product, the thermodynamic control of the intermediate led to the less thermodynamically favored thioester product. Further mechanistic studies revealed that the thioester formation followed a pathway where the thiol bonded and was subsequently dehydrogenated on a vacant site of the Ru catalyst. The insertion and subsequent dehydrogenation of the alcohol to the aldehyde was achieved in an outer sphere mechanism before the formation of the C–S bond and β–H elimination that led to the formation of the thioester and the regeneration of the catalyst (42).
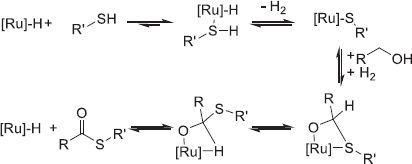 (42)
(42)
-
Conclusion
S-based LOHC exploration has been scarce due to the negative influence of sulphur on the catalysts and the rapid (de)hydrosulfuration of the structures producing toxic H2S. In addition, the atomic weight of the sulphur atom is detrimental to the gravimetric density of the LOHC system, hence O-containing LOHC was principally studied instead. Therefore, S-containing LOHC is of little interest if no catalytic system is able to efficiently answer these issues.
4.2.5.8 B,N-containing LOHC
The 1,2-dihydro-1,2-azaborine/1,2-BN-cylcohexane couple represents the frontier between ammonia-borane and the LOHC technologies which in theory can store up to 9.4 wt.%H2 if the system is completely dehydrogenated (43). From the ammonia-borane point of view, 1,2-BN-cylcohexane can undergo an intramolecular dehydrogenation on the B-N moiety and a trimerization process. On the LOHC side, the carbon atoms of the trimer can be dehydrogenated to form the analogous aromatic compound.
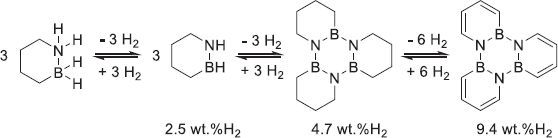 (43)
(43)
While these systems are air and moisture stable [540], 1,2-BN-cylcohexane is a solid that melts at 62–63 °C and is usually diluted in solvent like in a 35 wt.% THF solution, diminishing the gravimetric density to 3.3 wt.%H2 and volumetric density for the complete dehydrogenation [541].
-
Hydrogenation
The hydrogenation of the monocyclic aromatized structure was performed in two steps. Direct hydrogenation of the carbon atoms was achieved with a 25%Pd/C catalyst in 4 h at 80 °C and 3 bar H2, yielding the hydrogenated cycle in 99% yield by GCMS and 89% by NMR. Hydrogenation of the B and N atoms was more complicated and required hydride (KH) and acid (HCl) treatments respectively to afford their hydrogenated form in quantitative yields [542]. Trimer regeneration was performed by MeOH solvolysis treatment in 12 h at room temperature followed by the use of strong hydrides like LiAlH4 or BH3-THF to regenerate the boron atom in 47% and 71% yields from the trimer respectively [543]. Finally, the thermodynamic hydrogenation equilibrium at 80 °C and 10 bar H2 was calculated to 95% conversion of the starting material, indicating the relatively modest theoretical conditions required to perform the hydrogenation [541].
-
Dehydrogenation
The quantitative trimerization was originally observed at 150 °C in toluene for 2 h without any catalyst, for a 4.7 wt.%H2 density. No further dehydrogenation of the trimer to the aromatized structure was obtained [540]. Thermodynamic computational calculations showed an enthalpy of 32 kJ/mol H2 for the dehydrogenation of the monocyclic structure to its aromatized form, revealing the beneficial effect of the B–N bond on the thermodynamics of the system [542]. In addition, further thermodynamic calculations that the equilibrium conversion would be 99% for the dehydrogenated form under 1 bar H2 [541]. Further catalytic development revealed that CoCl2 was an excellent catalyst of the reaction that achieved the quantitative trimerization in 15 min at 80 °C in toluene. The neat dehydrogenation was also performed in the same conditions, but required in 4 h instead. The dehydrogenation of the cycles of the trimer was achieved with an Ir pincer catalyst in the presence of an H2 acceptor in mesithylene, reaching >99% conversion and 90% selectivity to the fully dehydrogenated product in 18 h at 160 °C. No attempts of the aromatic dehydrogenation of the trimer were reported without any H2 acceptor [543].
Finally, new generation 1,6;2,3-bis-BN-cyclohexane was proposed as a novel carrier system able to thermally release up to 9 wt.%H2 at 180 °C with only a slight decomposition of the carrier while the catalytic dehydrogenation with Pd/C released only 1.47 wt.%H2 at 50 °C [544].
-
Conclusion
B,N-containing LOHC interest is high due to their potential lower dehydrogenation enthalpy and high gravimetric density. Nevertheless, their stability and regeneration are key issues that still need to be addressed before B,N-LOHC can be industrially used.
4.2.5.9 Si-containing LOHC
Silanes in the presence of alcohol evolve H2, producing an alkoxysilane as a by-product. The potential best monosilane system is SiH4 with MeOH (5.0 wt.%H2) but SiH4 is a gas. To date, most studies were based on the PhMe2SiH-Methanol couple that possesses a very low gravimetric density of only 0.7 wt.%H2. It is worth noting that only the silane group participates in the storage/release of H2 and that the organic groups only tailor the release kinetics. Polysilanes have then been rapidly suggested as more efficient storage systems and tetrasilylmetane-methanol couple could potentially reach a gravimetric density of 8.82 wt.%H2 and 70 gH2/L (using the density of methanol) (44).
 (44)
(44)
Hysilabs, a French startup, is currently exploring this option with a purely inorganic silica-based liquid polymer, the Hydrosil. This new H2 carrier possesses an advantageous gravimetric density (8.7 wt.%H2) and is able to release H2 when mixed with water, producing heat as a byproduct. Compared to other classic LOHCs, this solution produces 40% less greenhouse gas emissions [545].
-
Hydrogenation
Much like alkoxy-boron (see 4.2.3), alkoxy-silane is highly stable, which poses a problem for their regeneration. While direct hydrogenation with heterogeneous catalysts is yet to be achieved, advances have been reported. Classic reducing agents like LiAlH4 [546] or borane species with sacrificial reagents like NaBH4 and ethylbromide [547] were originally used. New procedures are starting to emerge as shown by the regeneration of silyl triflates with 4 bar H2 and an Ir catalyst in the presence of a base, achieving 95% conversion at 60 °C in 48 h [548].
-
Dehydrogenation
Contrary to LOHC couples, the dehydrogenation is exothermic. The first dehydrogenation example of silanes dehydrogenation used water as a source of a proton with a heterogeneous Ag/hydroxyapatite catalyst that achieved complete conversion in 15 min [549]. However, the obtained Si–OH species were difficult to regenerate by classic methods, and Si–O–Si species were also created. H2 was also released in catalyst free conditions using silane and sodium methoxide at room temperature in 15 s [550]. Polysilanes and methanol coupling under Bu4NF activation achieved 100% conversion at 25 °C in 10 s [551]. The reaction activity was linked to the steric hindrance of the alcohol and diols could also be used on dihydrides silanes with yields superior to 90% at 60–90 °C [552]. Recent catalytic systems consisted of Ru [553] and Ir [554] complexes supported on rGO that could achieve superior conversion and stability on stream for 1 h [555]. Finally, an heterogeneous Cu-doped on ZIF-8 zeolite was proposed to achieve the complete dehydrogenation of PhMe2SiH, and in 9 h at 110 °C [556].
-
Conclusion
Silanes present high gravimetric densities, with an energy profile better suited for an intermittent energy scenario due to an exothermic dehydrogenation that can be performed at room temperature. However, their clean regeneration is not yet achieved, although recent progress with silyl triflates might pave the way for direct hydrogenation with H2. Future work could encompass the hydrogenation/dehydrogenation of the organic moiety to increase the storage capacity of the PhMe2SiH system as well as the study of inorganic liquids as H2 storage media.
4.2.5.10 Others
LOHC like N-ethylcarbazole can be turned into ionic liquids when linked to an imidazolium group by a 1 to 3 carbon chain. The addition of Si atoms in the chain allowed for the liquid state of the structure at room temperature. Hydrogenation and dehydrogenation experiments were performed on this ionic liquid with Pd/C and quantitative conversions and stability were observed up to 220 °C. However, the gravimetric densities were lowered to 2.05 wt.%H2 for the carbon linker, 1.58 wt.%H2 for the C–Si linker [557].
Finally, H2 can also be stored in frustrated-Lewis-pair-like benzimidazoline structures able to release H2 at temperatures as low as 80 °C using Pd(OH)2/C as presented in equation (45).
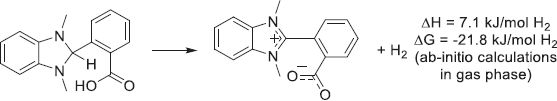 (45)
(45)
While this example demonstrated the possibility of creating organic hydride with exergonic properties, the system gravimetric density is very low (0.7 wt.%H2) and its reversibility has yet to be proven [558].
5. Conclusion and perspectives
Numerous H2 storage methods exist, but all have different drawbacks (energy, stability, synthetic accessibility, economics, etc.). However, a simple comparison of both gravimetric and volumetric densities of each chemical-based H2 storage system is pertinent to know at first glance which system could be in principle efficient for storing the highest H2 amounts (Fig. 23).
 |
Figure 23 Comparison of each chemical-based H2 storage system based on their gravimetric and volumetric H2 densities. The dotted lines represent the gravimetric and volumetric densities required by the US DOE for H2 storage. |
From a quick look, high gravimetric and volumetric densities materials are composed of metal hydrides, ammonia-borane, and circular hydrogen carriers while physical sorbents present reduced H2 storage properties. Comparatively, the LOHC (grey area) presents intermediate gravimetric (3-11 wt.%H2) and volumetric (38–96 gH2/L) densities which are sufficient to answer the US DOE criteria of 6.5 wt.%H2 and 62 gH2/L for vehicular transportation as long as the complete system is not much heavier than the LOHC capacity [559].
In addition, many other aspects must be taken into account in order to choose an appropriate H2 storage technology such as its cycling capacity, energy cost like the dehydrogenation enthalpy, reaction phase, catalysts, presence of solvents or additives, synthetic accessibility, cost, safety, transportability, environmental impact, produced H2 quality and flux and so on. These non-exhaustive secondary criteria are actually primordial and highly dependent on the targeted application. For example, the cycling capacity describes the durability of the LOHC, the dehydrogenation enthalpy its overall energy efficiency and the use of PGM-free heterogeneous catalysts usually limits the economic cost of the system. In addition, a liquid-phase reaction is advantageous over a gas-phase reaction as no expensive gas separation is required to obtain pure H2. Finally, following the 12 principles of green chemistry, reactions occurring without solvent or additives strongly reduce the toxicity profile and environmental impact of the process [560]. A comparison of these five secondary criteria for different LOHCs is presented in Table 1. Here, no state-of-the-art LOHC complies with all selected secondary criteria, indicating that the development of new LOHC couples is still necessary to overcome the presented barriers.
Comparison of different LOHC over various secondary criteria. Green signifies that the LOHC complies with the criterion, red that it does not.
Finally, the complexity of the industrial system must also be taken into account for further development, in particular the size and weight of the system, the network to collect and recharge H2 on the LOHC (containers, distribution, competition with already-in-use systems, etc.), economic costs and lastly the overall efficiency of the system.
Comparatively, to solid hydrogen storage systems like metal hydrides and ammonia-borane, the LOHC technology fares better on the transportability and economic aspects due to the possibility of using the current oil and gas infrastructures with only small modifications to the current system. In addition, due to their oil-like nature, LOHC also presents a better social acceptance for mobility options due to its similarity with the currently employed fuels [208]. Circular hydrogen carriers are serious contenders for the LOHC technology but are less attractive due to the gaseous nature of their hydrogen-lean molecules that induce gas separation and recovery at the gas exhaust or from the air. Therefore, these recent developments and environmental considerations promoted the LOHC technology as a mean to store vast quantities of energy (GWh to TWh ranges) for seasonal energy storage.
However, through this literature review, barriers for the current state-of-the-art LOHC systems were highlighted, in particular, their high dehydrogenation enthalpy, the rarity and cost of PGM used for the current hydrogenation and dehydrogenation catalysts, and finally the LOHC stability after multiple hydrogenation/dehydrogenation cycles. The current perspectives on each point are presented hereafter.
- 1.
Dehydrogenation enthalpy
The development of organic structures with novel reactivities following the reviewed literature such as C-N (e.g. N-Ethylcarbazole) and C–O (e.g. γ-Butyrolactone) bonds might provide low dehydrogenation enthalpy LOHC structures. Moreover, B–N and Si-based LOHC show high potential for new low-enthalpy carriers if their hydrogenation can be efficiently catalyzed. Finally, liquid inorganic H2 carriers (e.g. the Hydrosil) are also promising H2 storage media. Nevertheless, the development of a high throughput methodology to develop and test new LOHC and catalysts/supports is still required.
- 2.
PGM-free heterogeneous metal catalysts
Overall, the development of high kinetics non-PGM selective heterogeneous hydrogenation and dehydrogenation catalysts is required for the LOHC technology to be further implemented. The design and comprehension of the LOHC-metal-support interfaces in heterogeneous catalytic systems is also key as the objective of complete reaction selectivity might be unachievable if these interfaces cannot be rationally constituted and analyzed.
Moreover, as the metal catalyst activity is dependent on the LOHC structure, refinement of the LOHC molecular design could also be beneficial. Indeed, the development of C–O-based structures is particularly interesting as non-noble metal catalysts such as Cu can catalyze both hydrogenation and dehydrogenation reactions. As numerous C–O bond-containing molecules able to store/release H2 exist in nature, it is only a matter of time before their pivotal role as LOHC is recognized.
- 3.
LOHC stability
As complex structures tend to induce more side reactions, the simplification of the molecular systems might be efficient in lowering the formation of side products. However, structure simplification is also often synonymous with low-molecular-weight structures and accordingly lower boiling points, thus this method is rather limited if the dehydrogenation reaction is to occur in the liquid phase. Finally, a more realistic approach might be to observe which degradation structures with H2 storage capacity are formed during the cycling of the current state-of-the-art LOHC as they probably are the most stable LOHC structures for the currently available catalytic systems.
- 4.
H2-to-power setups and heat integration
Down the H2 chalue chain, the presence of degradation products in the H2 output could be detrimental for the H2-to-Power setups such as PEM-FC where membrane contamination will drastically shorten their life expectancy. SOE-FC are less sensitive to contaminants than PEM-FC, but their presence requires monitoring to ensure that no excessive degradation occurs. Conversely, thermal engines are however unaffected by traces of contaminants, but release greenhouse gases such as CO2 and NO x accordingly.
Heat integration from these conversion devices could in principle compensate the dehydrogenation enthalpy providing the dehydrogenation temperature is lower than their operating temperature [561]. Therefore, PEM-FC does not permit heat integration for the time being as the state-of-the-art LOHC dehydrogenation temperatures are 100–200 °C above their operating temperature. Conversely, SOE-FC is suitable for dehydrogenation heat integration. Nevertheless, their extreme operating temperature limits the application possibilities, especially for on-board systems. Finally, much like SOE-FC, thermal engines can be used to circumvent the dehydrogenation enthalpy cost but their extreme operating temperature produces NO x from air.
In conclusion, massive energy storage is a major concern due to the implementation of intermittent renewable energies in order to phase out fossil fuels out of our energy mix. Both must concentrate on research efforts to achieve a successful energy transition. While early studies targeted individual mobility as a LOHC application, the dehydrogenation temperature, limited H2 gravimetric and volumetric densities as well as the low cycling capacity of most current LOHC systems make this application unsuitable. Conversely, stationary systems for off-grid energy generation might be interesting if the system cost is low (LOHC molecules, catalysts, H2 production, hydrogenation, dehydrogenation, and H2-to-power setups) and the system volume might be less of an issue. Here, the degradation products could be used as either H2 storage materials of reduced capacity or fuel to compensate for the high dehydrogenation energy.
Nowadays, global massive energy storage and transportation is the most promising short-term application of the LOHC technology, but it would necessitate both a low system cost and high H2 densities. Here, the LOHC technology has a real pertinence to safely transport and distribute energy worldwide if the previously raised issues are answered. Indeed, it is worth noting that most of the system costs arise from the catalyst. Under the assumptions that H2 has already been produced, the complete system is available and heat integrated, and the yield for the conversion from H2 to electricity is 100%, a generation of 3 to 5 GWh over 1 h would require roughly 1400,000 to 2,400,000 m3 of hydrogenated LOHC 18H-DBT (6 to 10 M€) with 1.5–2.5 tons of Pt (45– 75 M€). While a more complete techno-economic study might reveal that another parameter of the system is more important than anticipated (H2 and energy costs, energy efficiency, etc.), these values show that using this technology as the only energy source might be impossible due to its high price. Therefore, a primary concern would be the reduction of the price of the catalyst if this technology is to compete with other H2 storage and transport technologies like Ammonia.
Acknowledgments
This work was supported by the CEA, the CNRS, the University Paris-Saclay, the European Research Council (ERC Consolidator Grant Agreement No. 818260), and the European project funding from the Fuel Cells and Hydrogen 2 Joint Undertaking under Grant Agreement No. 101007223.
References
- Ritchie H., Roser M., Rosado P. (2020) Energy, Our World in Data. [Google Scholar]
- Bruland K., Smith K. (2013) Assessing the role of steam power in the first industrial revolution: the early work of Nick von Tunzelmann, Res. Policy 42, 10, 1716–1723. https://doi.org/10.1016/j.respol.2012.12.008. [CrossRef] [Google Scholar]
- Clark G., Jacks D. (2007) Coal and the Industrial Revolution, 1700–1869, Eur. Rev. Econ. Hist. 11, 1, 39–72. https://doi.org/10.1017/S1361491606001870. [CrossRef] [Google Scholar]
- Clow A., Nan L.C. (1972) Vitriol in the industrial revolution, in Science, Technology and Economic Growth in the Eighteenth Century, Routledge. [Google Scholar]
- Nicolas Leblanc (1742-1806).Pdf. (accessed 2022-07-27) https://pubs.acs.org/doi/pdf/10.1021/ed019p567. [Google Scholar]
- Kanefsky J., Robey J. (1980) Steam engines in 18th-century Britain: a quantitative assessment, Technol. Cult. 21, 2, 161–186. https://doi.org/10.2307/3103337. [CrossRef] [Google Scholar]
- Alley R.B., Berntsen T., Bindoff N.L., Chen Z., Chidthaisong A., Friedlingstein P., Hegerl G.C., Heimann M., Hewitson B., Hoskins B.J., Joos F., Jouzel J., Kattsov V., Lohmann U., Manning M., Matsuno T., Molina M., Nicholls N., Overpeck J., Qin D., Raga G., Ramaswamy V., Ren J., Rusticucci M., Solomon S., Somerville R., Stocker T.F., Stott A., Stouffer R.J., Whetton P., Wood R.A., Wratt D., Arblaster J., Brasseur G., Christensen J.H., Denman K.L., Fahey D.W., Forster P., Jansen E., Jones P.D., Knutti R., Treut H.L., Lemke P., Meehl G., Mote P., Randall D.A., Stone D.A., Trenberth K.E., Willebrand J., Zwiers F.. IPCC Sixth Assessment, summary for Policymakers, p. 18. [Google Scholar]
- IPCC (2006) IPCC Guidelines for National Greenhouse Gas Inventories, Volume 1, General Guidance and Reporting (accessed 2021-01-20). https://www.ipcc-nggip.iges.or.jp/public/2006gl/vol1.html. [Google Scholar]
- Karl T.R., Trenberth K.E. (2003) Modern global climate change, Science 302, 5651, 1719–1723. https://doi.org/10.1126/science.1090228. [CrossRef] [PubMed] [Google Scholar]
- Montzka S.A., Dlugokencky E.J., Butler J.H. (2011) Non-CO2 greenhouse gases and climate change, Nature 476, 7358, 43–50. https://doi.org/10.1038/nature10322. [CrossRef] [PubMed] [Google Scholar]
- IPCC. IPCC_AR6_WGII_SummaryForPolicymakers.Pdf (accessed 2022-07-13) https://report.ipcc.ch/ar6wg2/pdf/IPCC_AR6_WGII_SummaryForPolicymakers.pdf [Google Scholar]
- Our World in Data. The world’s energy problem. Our World in Data. (accessed 2022-07-13). https://ourworldindata.org/worlds-energy-problem [Google Scholar]
- Gibon T., Arvesen A., Hertwich E.G. (2017) Life cycle assessment demonstrates environmental co-benefits and trade-offs of low-carbon electricity supply options, Renew. Sust. Energ. Rev. 76, 1283–1290. https://doi.org/10.1016/j.rser.2017.03.078. [CrossRef] [Google Scholar]
- Hondo H. (2005) Life cycle GHG emission analysis of power generation systems: Japanese case, Energy 30, 11, 2042–2056. https://doi.org/10.1016/j.energy.2004.07.020. [CrossRef] [Google Scholar]
- Amponsah N.Y., Troldborg M., Kington B., Aalders I., Hough R.L. (2014) Greenhouse gas emissions from renewable energy sources: a review of lifecycle considerations, Renew. Sust. Energ. Rev. 39, 461–475. https://doi.org/10.1016/j.rser.2014.07.087. [CrossRef] [Google Scholar]
- Chiffres clés de l’énergie – Édition 2019, p. 80. [Google Scholar]
- Global Electricity Review (2022) Ember (accessed 2022-07-27) https://ember-climate.org/insights/research/global-electricity-review-2022/ [Google Scholar]
- LOI N° 2015-992 Du 17 Août 2015 Relative à La Transition Énergétique Pour La Croissance Verte (2015). [Google Scholar]
- Bremen L.V. (2010) Large-scale variability of weather dependent renewable energy sources, in Management of Weather and Climate Risk in the Energy Industry, A. Troccoli (ed.), NATO Science for Peace and Security Series C: Environmental Security. Dordrecht, Springer, Netherlands, pp. 189–206. https://doi.org/10.1007/978-90-481-3692-6_13 [CrossRef] [Google Scholar]
- éCO2mix - La production d’électricité par filière (accessed 2022-07-27). https://www.rte-france.com/eco2mix/la-production-delectricite-par-filiere [Google Scholar]
- Revol, M. Quand trop d’énergies renouvelables privent la Californie… d’électricité. Le Point (accessed 2020-08-25). https://www.lepoint.fr/economie/quand-trop-d-energies-renouvelables-privent-la-californie-d-electricite-20-08-2020-2388408_28.php. [Google Scholar]
- Energy Charts (accessed 2021-01-21). https://energy-charts.info/?l=fr&c=DE [Google Scholar]
- Saboori H., Hemmati R., Ghiasi S.M.S., Dehghan S. (2017) Energy storage planning in electric power distribution networks – a state-of-the-art review, Renew. Sust. Energ. Rev. 79, 1108–1121. https://doi.org/10.1016/j.rser.2017.05.171. [CrossRef] [Google Scholar]
- Akinyele D.O., Rayudu R.K. (2014) Review of energy storage technologies for sustainable power networks, Sustain. Energy Technol. Assess. 8, 74–91. https://doi.org/10.1016/j.seta.2014.07.004. [Google Scholar]
- Orecchini F. (2006) The era of energy vectors, Int. J. Hydrogen Energy 31, 14, 1951–1954. https://doi.org/10.1016/j.ijhydene.2006.01.015. [CrossRef] [Google Scholar]
- Chen L., Zheng T., Mei S., Xue X., Liu B., Lu Q. (2016) Review and prospect of compressed air energy storage system, J. Mod. Power Syst. Clean Energy 4, 4, 529–541. https://doi.org/10.1007/s40565-016-0240-5. [CrossRef] [Google Scholar]
- Geth F., Brijs T., Kathan J., Driesen J., Belmans R. (2015) An overview of large-scale stationary electricity storage plants in Europe: current status and new developments, Renew. Sust. Energ. Rev. 52, 1212–1227. https://doi.org/10.1016/j.rser.2015.07.145. [CrossRef] [Google Scholar]
- Capacitor-with-Cover-Page-v2.Pdf (accessed 2022-07-29). https://d1wqtxts1xzle7.cloudfront.net/37529848/capacitor-with-cover-page-v2.pdf [Google Scholar]
- Kebede A.A., Kalogiannis T., Van Mierlo J., Berecibar M. (2022) A comprehensive review of stationary energy storage devices for large scale renewable energy sources grid integration, Renew. Sust. Energ. Rev. 159, 112213, https://doi.org/10.1016/j.rser.2022.112213. [CrossRef] [Google Scholar]
- Park K., Zhang Z. (2013) Fundamentals and applications of near-field radiative energy transfer, Front. Heat Mass Transf. 4, 1, 013001. [CrossRef] [Google Scholar]
- Duigou Le (2000) A. La filière hydrogène Un moyen de stockage de l’énergie (accessed 2020-08-25). https://hal.archives-ouvertes.fr/hal-02416323/file/201500004533.pdf [Google Scholar]
- Momirlan M., Veziroglu T.N. (2005) The properties of hydrogen as fuel tomorrow in sustainable energy system for a cleaner planet, Int. J. Hydrogen Energy 30, 7, 795–802. https://doi.org/10.1016/j.ijhydene.2004.10.011. [CrossRef] [Google Scholar]
- Abbasi T., Abbasi S.A. (2011) “Renewable” hydrogen: prospects and challenges, Renew. Sust. Energ. Rev. 15, 6, 3034–3040. https://doi.org/10.1016/j.rser.2011.02.026. [CrossRef] [Google Scholar]
- Holbrook J.H., Cialone H.J., Collings E.W., Drauglis E.J., Scott P.M., Mayfield M.E. (2012) 5 – Control of hydrogen embrittlement of metals by chemical inhibitors and coatings, in: Gaseous Hydrogen Embrittlement of Materials in Energy Technologies, Vol. 1, R.P. Gangloff, B.P. Somerday (eds.), Woodhead Publishing Series in Metals and Surface Engineering. Woodhead Publishing, pp. 129–153. https://doi.org/10.1533/9780857095374.1.129. [CrossRef] [Google Scholar]
- Holbrook J.H., Cialone H.J., Scott P.M. (1984) Hydrogen Degradation of Pipeline Steels. Summary Report, Battelle Columbus Labs., BNL-51855, OH, USA (accessed 2020-03-04). https://www.osti.gov/biblio/5985541 [CrossRef] [Google Scholar]
- Veziroglu T.N. (2016) Metal-Hydrogen Systems: Proceedings of the Miami International Symposium on Metal-Hydrogen Systems, 13–15 April 1981, Miami Beach, Florida, USA, Elsevier. [Google Scholar]
- Gupta R.B. (2008) Hydrogen fuel: production, transport, and storage, CRC Press. [CrossRef] [Google Scholar]
- IEA. The Future of Hydrogen – Analysis. IEA (accessed 2021-01-22). https://www.iea.org/reports/the-future-of-hydrogen [Google Scholar]
- Christensen H., Bjergbakke E. (1982) Radiolysis of ground water from spent fuel. SKBF-KBS-TR–82-18, Svensk Kaernbraenslefoersoerjning AB (accessed 2020-06-30) http://inis.iaea.org/Search/search.aspx?orig_q=RN:14788675 [Google Scholar]
- Prinzhofer A., Tahara Cissé C.S., Diallo A.B. (2018) Discovery of a large accumulation of natural hydrogen in Bourakebougou (Mali), Int. J. Hydrogen Energy 43, 42, 19315–19326. https://doi.org/10.1016/j.ijhydene.2018.08.193. [CrossRef] [Google Scholar]
- Stevens T.O., McKinley J.P. (2000) Abiotic controls on H2 production from Basalt−Water reactions and implications for aquifer biogeochemistry, Environ. Sci. Technol. 34, 5, 826–831. https://doi.org/10.1021/es990583g. [CrossRef] [Google Scholar]
- Hosseini S.E., Wahid M.A. (2016) Hydrogen production from renewable and sustainable energy resources: promising green energy carrier for clean development, Renew. Sust. Energ. Rev. 57, 850–866. https://doi.org/10.1016/j.rser.2015.12.112. [CrossRef] [Google Scholar]
- Cao L., Yu I.K.M., Xiong X., Tsang D.C.W., Zhang S., Clark J.H., Hu C., Ng Y.H., Shang J., Ok Y.S. (2020) Biorenewable hydrogen production through biomass gasification: a review and future prospects, Environ. Res. 186, 109547. https://doi.org/10.1016/j.envres.2020.109547. [CrossRef] [Google Scholar]
- Sengodan S., Lan R., Humphreys J., Du D., Xu W., Wang H., Tao S. (2018) Advances in reforming and partial oxidation of hydrocarbons for hydrogen production and fuel cell applications, Renew. Sust. Energ. Rev. 82, 761–780. https://doi.org/10.1016/j.rser.2017.09.071. [CrossRef] [Google Scholar]
- Kothari R., Buddhi D., Sawhney R.L. (2008) Comparison of environmental and economic aspects of various hydrogen production methods, Renew. Sust. Energ. Rev. 12, 2, 553–563. https://doi.org/10.1016/j.rser.2006.07.012. [CrossRef] [Google Scholar]
- Baruah R., Dixit M., Basarkar P., Parikh D., Bhargav A. (2015) Advances in ethanol autothermal reforming, Renew. Sust. Energ. Rev. 51, 1345–1353. https://doi.org/10.1016/j.rser.2015.07.060. [CrossRef] [Google Scholar]
- da Silva Veras T., Mozer T.S., da Costa Rubim Messeder dos Santos D., da Silva César A. (2017) Hydrogen: trends, production and characterization of the main process worldwide, Int. J. Hydrogen Energy 42, 4, 2018–2033. https://doi.org/10.1016/j.ijhydene.2016.08.219.. [CrossRef] [Google Scholar]
- Muradov N.Z. (1993) How to produce hydrogen from fossil fuels without CO2 emission, Int. J. Hydrogen Energy 18, 3, 211–215. https://doi.org/10.1016/0360-3199(93)90021-2. [CrossRef] [Google Scholar]
- Methane splitting and turquoise ammonia – Ammonia Energy Association (accessed 2022-08-03). https://www.ammoniaenergy.org/articles/methane-splitting-and-turquoise-ammonia/ [Google Scholar]
- Machhammer O., Bode A., Hormuth W. (2016) Financial and ecological evaluation of hydrogen production processes on large scale, Chem. Eng. Technol. 39, 6, 1185–1193. https://doi.org/10.1002/ceat.201600023. [CrossRef] [Google Scholar]
- 1 Scale up BASF.Pdf (accessed 2022-08-03). https://arpa-e.energy.gov/sites/default/files/1%20Scale%20up%20BASF.pdf. [Google Scholar]
- Keller M. (2021) Comment on “Methane pyrolysis for zero-emission hydrogen production: a potential bridge technology from fossil fuels to a renewable and sustainable hydrogen economy”, Ind. Eng. Chem. Res. 60, 48, 17792–17794. https://doi.org/10.1021/acs.iecr.1c03926. [CrossRef] [Google Scholar]
- Chisholm G., Cronin L. (2016) Chapter 16 – Hydrogen from water electrolysis, in Storing Energy, T.M. Letcher (ed.), Oxford, Elsevier, pp. 315–343. https://doi.org/10.1016/B978-0-12-803440-8.00016-6 [CrossRef] [Google Scholar]
- Smolinka T., Bergmann H., Garche J., Kusnezoff M. (2022) Chapter 4 – The history of water electrolysis from its beginnings to the present, in Electrochemical Power Sources: Fundamentals, Systems, and Applications, T. Smolinka, J. Garche (eds.), Elsevier, pp. 83–164. https://doi.org/10.1016/B978-0-12-819424-9.00010-0 [CrossRef] [Google Scholar]
- Ursua A., Gandia L.M., Sanchis P. (2012) Hydrogen production from water electrolysis: current status and future trends, Proc. IEEE 100, 2, 410–426. https://doi.org/10.1109/JPROC.2011.2156750. [CrossRef] [Google Scholar]
- IEA. Electrolysers – Analysis. IEA (accessed 2023-01-11). https://www.iea.org/reports/electrolysers [Google Scholar]
- Zeng K., Zhang D. (2010) Recent progress in alkaline water electrolysis for hydrogen production and applications, Prog. Energy Combust. Sci. 36, 3, 307–326. https://doi.org/10.1016/j.pecs.2009.11.002. [CrossRef] [Google Scholar]
- Sahu A.K., Pitchumani S., Sridhar P., Shukla A.K. (2009) Nafion and modified-Nafion membranes for polymer electrolyte fuel cells: an overview, Bull. Mater Sci. 32, 3, 285–294. https://doi.org/10.1007/s12034-009-0042-8. [CrossRef] [Google Scholar]
- Vincent I., Bessarabov D. (2018) Low cost hydrogen production by anion exchange membrane electrolysis: a review, Renew. Sust. Energ. Rev. 81, 1690–1704. https://doi.org/10.1016/j.rser.2017.05.258. [CrossRef] [Google Scholar]
- Bockris J.O’M., Conway B.E., Yeager E., White R.E. (1981) Electrochemical processing, in: Comprehensive Treatise of Electrochemistry, Vol. 2, Plenum Press, New York, London. BBPCAX. Berichte der Bunsengesellschaft für physikalische Chemie. Ber. Bunsenges. Phys. Chem. 1982, 86 (6), 575–576. https://doi.org/10.1002/bbpc.19820860631 [Google Scholar]
- Holladay J.D., Hu J., King D.L., Wang Y. (2009) An overview of hydrogen production technologies, Catal. Today 139, 4, 244–260. https://doi.org/10.1016/j.cattod.2008.08.039. [CrossRef] [Google Scholar]
- Nechache A., Hody S. (2021) Alternative and innovative solid oxide electrolysis cell materials: a short review, Renew. Sust. Energ. Rev. 149, 111322. https://doi.org/10.1016/j.rser.2021.111322. [CrossRef] [Google Scholar]
- Seitz M., von Storch H., Nechache A., Bauer D. (2017) Techno economic design of a solid oxide electrolysis system with solar thermal steam supply and thermal energy storage for the generation of renewable hydrogen, Int. J. Hydrogen Energy 42, 42, 26192–26202. https://doi.org/10.1016/j.ijhydene.2017.08.192. [CrossRef] [Google Scholar]
- Zheng H., Sullivan C., Mereddy R., Zeng R., Duke M., Clarke W. (2021) Production of bio-hydrogen using a membrane anaerobic reactor: limitations due to diffusion. [Google Scholar]
- Lee H.-S., Salerno M.B., Rittmann B.E. (2008) Thermodynamic evaluation on H2 production in glucose fermentation, Environ. Sci. Technol. 42, 7, 2401–2407. https://doi.org/10.1021/es702610v. [CrossRef] [PubMed] [Google Scholar]
- Bshish A., Yaakob Z., Narayanan B., Ramakrishnan R., Ebshish A. (2011) Steam-reforming of ethanol for hydrogen production, Chemical Papers 65, 3, 251–266. https://doi.org/10.2478/s11696-010-0100-0. [CrossRef] [Google Scholar]
- Fahmy T.Y.A., Fahmy Y., Mobarak F., El-Sakhawy M., Abou-Zeid R.E. (2020) Biomass pyrolysis: past, present, and future, Environ. Dev. Sustain. 22, 1, 17–32. https://doi.org/10.1007/s10668-018-0200-5. [CrossRef] [Google Scholar]
- Demirbas A. (2008) Hydrogen production from carbonaceous solid wastes by steam reforming, Energy Sources A: Recovery Util. Environ. Eff. 30, 10, 924–931. https://doi.org/10.1080/10826070601082658. [CrossRef] [Google Scholar]
- Calzavara Y., Joussot-Dubien C., Boissonnet G., Sarrade S. (2005) Evaluation of biomass gasification in supercritical water process for hydrogen production, Energy Convers. Manage. 46, 4, 615–631. https://doi.org/10.1016/j.enconman.2004.04.003. [CrossRef] [Google Scholar]
- Oni A.O., Anaya K., Giwa T., Di Lullo G., Kumar A. (2022) Comparative assessment of blue hydrogen from steam methane reforming, autothermal reforming, and natural gas decomposition technologies for natural gas-producing regions, Energy Convers. Manage. 254, 115245. https://doi.org/10.1016/j.enconman.2022.115245. [CrossRef] [Google Scholar]
- Bartels J.R., Pate M.B., Olson N.K. (2010) An economic survey of hydrogen production from conventional and alternative energy sources, Int. J. Hydrogen Energy 35, 16, 8371–8384. https://doi.org/10.1016/j.ijhydene.2010.04.035. [CrossRef] [Google Scholar]
- Kreutz T., Williams R., Consonni S., Chiesa P. (2005) Co-production of hydrogen, electricity and CO2 from coal with commercially ready technology. Part B: economic analysis, Int. J. Hydrogen Energy 30, 7, 769–784. https://doi.org/10.1016/j.ijhydene.2004.08.001. [CrossRef] [Google Scholar]
- Simbeck D.R. (2005) Hydrogen costs with CO2 capture, in Greenhouse Gas Control Technologies, Vol. 7, E.S. Rubin, D.W. Keith, C.F. Gilboy, M. Wilson, T. Morris, J. Gale, K. Thambimuthu (eds.), Elsevier Science Ltd, Oxford, pp. 1059–1066. https://doi.org/10.1016/B978-008044704-9/50108-7. [CrossRef] [Google Scholar]
- Damen K., van Troost M., Faaij A., Turkenburg W. (2006) A comparison of electricity and hydrogen production systems with CO2 capture and storage. Part A: Review and selection of promising conversion and capture technologies, Prog. Energy Combust. Sci. 32, 2, 215–246. https://doi.org/10.1016/j.pecs.2005.11.005. [CrossRef] [Google Scholar]
- Levene J., Kroposki B., Sverdrup G. (2006) Wind energy and production of hydrogen and electricity – opportunities for renewable hydrogen, in: Preprint: 2006 POWER-GEN Renewable Energy and Fuels Technical Conference, Las Vegas, Nevada, p. 18. [Google Scholar]
- Olateju B., Kumar A. (2016) A techno-economic assessment of hydrogen production from hydropower in Western Canada for the upgrading of bitumen from oil sands, Energy 115, 604–614. https://doi.org/10.1016/j.energy.2016.08.101. [CrossRef] [Google Scholar]
- El-Emam R.S., Özcan H. (2019) Comprehensive review on the techno-economics of sustainable large-scale clean hydrogen production, J. Clean. Prod. 220, 593–609. https://doi.org/10.1016/j.jclepro.2019.01.309. [CrossRef] [Google Scholar]
- Padro C.E.G., Putsche V. (1999) Survey of the economics of hydrogen technologies, Technical Report. NREL/TP-570-27079 (NREL), 12212, National Renewable Energy Lab, Golden, CO (United States). https://doi.org/https://doi.org/10.2172/12212. [CrossRef] [Google Scholar]
- Sathyaprakasan P., Kannan G. (2015) BITS Pilani-Dubai Campus Dubai. Economics of bio-hydrogen production, IJESD 6, 4, 352–356. https://doi.org/10.7763/IJESD.2015.V6.617. [CrossRef] [Google Scholar]
- Al-Qahtani A., Parkinson B., Hellgardt K., Shah N., Guillen-Gosalbez G. (2021) Uncovering the true cost of hydrogen production routes using life cycle monetisation, Appl. Energy 281, 115958. https://doi.org/10.1016/j.apenergy.2020.115958. [CrossRef] [Google Scholar]
- Air Liquide (2023) Encyclopédie des gaz Air Liquide, Gas Encyclopedia (accessed 2022-08-05) https://encyclopedia.airliquide.com/fr [Google Scholar]
- von Helmolt R., Eberle U. (2007) Fuel cell vehicles: status 2007, J. Power Sourc. 165, 2, 833–843. https://doi.org/10.1016/j.jpowsour.2006.12.073. [CrossRef] [Google Scholar]
- Pasman H.J., Rogers W.J. (2012) Risk assessment by means of bayesian networks: a comparative study of compressed and liquefied H2 transportation and tank station risks, Int. J. Hydrogen Energy 37, 22, 17415–17425. https://doi.org/10.1016/j.ijhydene.2012.04.051. [CrossRef] [Google Scholar]
- Hua T.Q., Ahluwalia R.K., Peng J.-K., Kromer M., Lasher S., McKenney K., Law K., Sinha J. (2011) Technical assessment of compressed hydrogen storage tank systems for automotive applications, Int. J. Hydrogen Energy 36, 4, 3037–3049. https://doi.org/10.1016/j.ijhydene.2010.11.090. [CrossRef] [Google Scholar]
- Parks G., Boyd R., Cornish J., Remick R. (2014) Hydrogen station compression, storage, and dispensing technical status and costs: systems integration, National Renewable Energy Lab (NREL), United States. https://doi.org/10.2172/1130621 [Google Scholar]
- Air Liquide Energies (2023) Comment stocker l’hydrogène ? Air Liquide Energies (accessed 2022-08-05). https://energies.airliquide.com/fr/mediatheque-planete-hydrogene/comment-stocker-lhydrogene [Google Scholar]
- Aasadnia M., Mehrpooya M. (2018) Large-scale liquid hydrogen production methods and approaches: a review, Appl. Energy 212, 57–83. https://doi.org/10.1016/j.apenergy.2017.12.033. [CrossRef] [Google Scholar]
- Bliesner R.M. (2013) Parahydrogen-orthohydrogen conversion for boil-off reduction from space stage fuel systems. Masters of Science in Mechanical Engineering, Washington State University, p. 49. [Google Scholar]
- Tzimas E., Filiou C., Peteves S., Veyret J. (2003) Hydrogen storage: state-of-the-art and future perspective, EUR 20995 EN. Cat. No. LD-NA-20995-EN-C; 2003. JRC26493. [Google Scholar]
- Makridis S. (2016) Hydrogen storage and compression, Chem. Phys. 1–28. https://doi.org/10.1049/PBPO101E_ch1. [Google Scholar]
- Peschka W. (1984) Liquid hydrogen as a vehicular fuel – a challenge for cryogenic engineering, Int. J. Hydrogen Energy 9, 6, 515–523. https://doi.org/10.1016/0360-3199(84)90104-6. [CrossRef] [Google Scholar]
- Stewart W.F. (1984) Operating experience with a liquid-hydrogen fueled buick and refueling system, Int. J. Hydrogen Energy 9, 6, 525–538. https://doi.org/10.1016/0360-3199(84)90105-8. [CrossRef] [Google Scholar]
- Furuhama S., Kobayashi Y. (1984) Development of a Hot-Surface-Ignition Hydrogen Injection Two-Stroke Engine, Int. J. Hydrogen Energy 9, 3, 205–213. https://doi.org/10.1016/0360-3199(84)90120-4. [CrossRef] [Google Scholar]
- Hassannayebi N., Azizmohammadi S., De Lucia M., Ott H. (2019) Underground hydrogen storage: application of geochemical modelling in a case study in the Molasse Basin, Upper Austria, Environ. Earth Sci. 78, 5, 177. https://doi.org/10.1007/s12665-019-8184-5. [CrossRef] [Google Scholar]
- Zivar D., Kumar S., Foroozesh J. (2021) Underground hydrogen storage: a comprehensive review, Int. J. Hydrogen Energy 46, 45, 23436–23462. https://doi.org/10.1016/j.ijhydene.2020.08.138. [CrossRef] [Google Scholar]
- Michalski J., Bünger U., Crotogino F., Donadei S., Schneider G.-S., Pregger T., Cao K.-K., Heide D. (2017) Hydrogen generation by electrolysis and storage in salt caverns: potentials, economics and systems aspects with regard to the German energy transition, Int. J. Hydrogen Energy 42, 19, 13427–13443. https://doi.org/10.1016/j.ijhydene.2017.02.102. [CrossRef] [Google Scholar]
- Commissariat général au développement durable (2021) Bilan énergétique de la France, Chiffres clés de l’énergie – Édition 2021 (accessed 2022-08-10). https://www.statistiques.developpement-durable.gouv.fr/edition-numerique/chiffres-cles-energie-2021/6-bilan-energetique-de-la-france.php [Google Scholar]
- Zhou L. (2005) Progress and problems in hydrogen storage methods, Renew. Sust. Energ. Rev. 9, 4, 395–408. https://doi.org/10.1016/j.rser.2004.05.005. [CrossRef] [Google Scholar]
- Ansón A., Benham M., Jagiello J., Callejas M.A., Benito A.M., Maser W.K., Züttel A., Sudan P., Martínez M.T. (2004) Hydrogen adsorption on a single-walled carbon nanotube material: a comparative study of three different adsorption techniques, Nanotechnology 15, 11, 1503–1508. https://doi.org/10.1088/0957-4484/15/11/023. [CrossRef] [Google Scholar]
- Zhou L., Zhou Y., Sun Y. (2004) Enhanced storage of hydrogen at the temperature of liquid nitrogen, Int. J. Hydrogen Energy 29, 3, 319–322. https://doi.org/10.1016/S0360-3199(03)00155-1. [CrossRef] [Google Scholar]
- Chen P., Wu X., Lin J., Tan K.L. (1999) High H2 uptake by alkali-doped carbon nanotubes under ambient pressure and moderate temperatures, Science 285, 5424, 91–93. https://doi.org/10.1126/science.285.5424.91. [CrossRef] [PubMed] [Google Scholar]
- Yang R.T. (2000) Hydrogen storage by alkali-doped carbon nanotubes – revisited, Carbon 38, 4, 623–626. https://doi.org/10.1016/S0008-6223(99)00273-0. [CrossRef] [Google Scholar]
- Tibbetts G.G., Meisner G.P., Olk C.H. (2001) Hydrogen storage capacity of carbon nanotubes, filaments, and vapor-grown fibers, Carbon 39, 15, 2291–2301. https://doi.org/10.1016/S0008-6223(01)00051-3. [CrossRef] [Google Scholar]
- Liu C., Chen Y., Wu C.-Z., Xu S.-T., Cheng H.-M. (2010) Hydrogen Storage in Carbon Nanotubes Revisited, Carbon 48 2, 452–455. https://doi.org/10.1016/j.carbon.2009.09.060. [CrossRef] [Google Scholar]
- Zhao W., Fierro V., Fernández-Huerta N., Izquierdo M.T., Celzard A. (2012) Impact of synthesis conditions of KOH activated carbons on their hydrogen storage capacities, Int. J. Hydrogen Energy 37, 19, 14278–14284. https://doi.org/10.1016/j.ijhydene.2012.06.110. [CrossRef] [Google Scholar]
- Kolesnikov A.I., Antonov V.E., Bashkin I.O., Li J.C., Moravsky A.P., Ponyatovsky E.G., Tomkinson J. (1999) Neutron spectroscopy of fullerite hydrogenated under high pressures, Phys. B Condens. Matter 263–264, 436–438. https://doi.org/10.1016/S0921-4526(98)01403-3. [CrossRef] [Google Scholar]
- Liu C., Fan Y.Y., Liu M., Cong H.T., Cheng H.M., Dresselhaus M.S. (1999) Hydrogen storage in single-walled carbon nanotubes at room temperature, Science 286, 5442, 1127–1129. https://doi.org/10.1126/science.286.5442.1127. [CrossRef] [PubMed] [Google Scholar]
- Ramimoghadam D., Gray E.M., Webb C.J. (2016) Review of polymers of intrinsic microporosity for hydrogen storage applications, Int. J. Hydrogen Energy 41, 38, 16944–16965. https://doi.org/10.1016/j.ijhydene.2016.07.134. [CrossRef] [Google Scholar]
- McKeown N.B., Gahnem B., Msayib K.J., Budd P.M., Tattershall C.E., Mahmood K., Tan S., Book D., Langmi H.W., Walton A. (2006) Towards polymer-based hydrogen storage materials: engineering ultramicroporous cavities within polymers of intrinsic microporosity, Angewandte Chemie 118, 11, 1836–1839. https://doi.org/10.1002/ange.200504241. [CrossRef] [Google Scholar]
- Tian M., Rochat S., Polak-Kraśna K., Holyfield L.T., Burrows A.D., Bowen C.R., Mays T.J. (2019) Nanoporous polymer-based composites for enhanced hydrogen storage, Adsorption 25, 4, 889–901. https://doi.org/10.1007/s10450-019-00065-x. [CrossRef] [Google Scholar]
- Côté A.P., Benin A.I., Ockwig N.W., O’Keeffe M., Matzger A.J., Yaghi O.M. (2005) Porous, crystalline, covalent organic frameworks, Science 310, 5751, 1166–1170. https://doi.org/10.1126/science.1120411. [CrossRef] [PubMed] [Google Scholar]
- Freund R., Zaremba O., Arnauts G., Ameloot R., Skorupskii G., Dincă M., Bavykina A., Gascon J., Ejsmont A., Goscianska J., Kalmutzki M., Lächelt U., Ploetz E., Diercks C.S., Wuttke S. (2021) The current status of MOF and COF applications, Angew. Chem. Int. Ed. 60, 45, 23975–24001. https://doi.org/10.1002/anie.202106259. [CrossRef] [PubMed] [Google Scholar]
- Han S.S., Furukawa H., Yaghi O.M., Goddard W.A. (2008) Covalent organic frameworks as exceptional hydrogen storage materials, J. Am. Chem. Soc. 130, 35, 11580–11581. https://doi.org/10.1021/ja803247y. [CrossRef] [PubMed] [Google Scholar]
- Klontzas E., Tylianakis E., Froudakis G.E. (2010) Designing 3D COFs with enhanced hydrogen storage capacity, Nano Lett. 10, 2, 452–454. https://doi.org/10.1021/nl903068a. [CrossRef] [PubMed] [Google Scholar]
- Pramudya Y., Mendoza-Cortes J.L. (2016) Design principles for high H2 storage using chelation of abundant transition metals in covalent organic frameworks for 0–700 Bar at 298 K, J. Am. Chem. Soc. 138, 46, 15204–15213. https://doi.org/10.1021/jacs.6b08803. [CrossRef] [PubMed] [Google Scholar]
- Shet S.P., Shanmuga Priya S., Sudhakar K., Tahir M. (2021) A review on current trends in potential use of metal-organic framework for hydrogen storage, Int. J. Hydrogen Energy 46, 21, 11782–11803. https://doi.org/10.1016/j.ijhydene.2021.01.020. [CrossRef] [Google Scholar]
- Grünker R., Bon V., Müller P., Stoeck U., Krause S., Mueller U., Senkovska I., Kaskel S. (2014) A New Metal-Organic Framework with Ultra-High Surface Area, Chem. Commun. 50, 26, 3450–3452. https://doi.org/10.1039/C4CC00113C. [CrossRef] [PubMed] [Google Scholar]
- Campesi R., Cuevas F., Latroche M., Hirscher M. (2010) Hydrogen spillover measurements of unbridged and bridged metal-organic frameworks – revisited, Phys. Chem. Chem. Phys. 12, 35, 10457–10459. https://doi.org/10.1039/C0CP00037J. [CrossRef] [PubMed] [Google Scholar]
- Luzan S.M., Talyzin A.V. (2010) Hydrogen adsorption in Pt catalyst/MOF-5 materials, Microporous Mesoporous Mater. 135, 1, 201–205. https://doi.org/10.1016/j.micromeso.2010.07.018. [CrossRef] [Google Scholar]
- Prins R. (2012) Hydrogen spillover. Facts and fiction, Chem. Rev. 112, 5, 2714–2738. https://doi.org/10.1021/cr200346z. [CrossRef] [PubMed] [Google Scholar]
- Barrer R.M. (1978) Zeolites and clay minerals as sorbents and molecular sieves, Academic Press (accessed 2022-08-23). https://scholar.google.com/scholar_lookup?title=Zeolites+and+clay+minerals+as+sorbents+and+molecular+sieves&author=Barrer%2C+R.+M.+%28Richard+Maling%29&publication_year=1978 [Google Scholar]
- Fraenkel D., Shabtai J. (1977) Encapsulation of hydrogen in molecular sieve zeolites, J. Am. Chem. Soc. 99, 21, 7074–7076. https://doi.org/10.1021/ja00463a058. [CrossRef] [Google Scholar]
- Weitkamp J., Fritz M., Ernst S. (1995) Zeolites as media for hydrogen storage, Int. J. Hydrogen Energy 20, 12, 967–970. https://doi.org/10.1016/0360-3199(95)00058-L. [CrossRef] [Google Scholar]
- Nishimiya N., Kishi T., Mizushima T., Matsumoto A., Tsutsumi K. (2001) Hyperstoichiometric hydrogen occlusion by palladium nanoparticles included in NaY zeolite, J. Alloys Compd. 319, 1, 312–321. https://doi.org/10.1016/S0925-8388(01)00921-5. [CrossRef] [Google Scholar]
- Langmi H.W., Book D., Walton A., Johnson S.R., Al-Mamouri M.M., Speight J.D., Edwards P.P., Harris I.R., Anderson P.A. (2005) Hydrogen storage in ion-exchanged zeolites, J. Alloys Compd. 404–406, 637–642. https://doi.org/10.1016/j.jallcom.2004.12.193. [CrossRef] [Google Scholar]
- Vitillo J.G., Ricchiardi G., Spoto G., Zecchina A. (2005) Theoretical maximal storage of hydrogen in zeolitic frameworks, Phys. Chem. Chem. Phys. 7, 23, 3948–3954. https://doi.org/10.1039/B510989B. [CrossRef] [PubMed] [Google Scholar]
- Veluswamy H.P., Kumar R., Linga P. (2014) Hydrogen storage in clathrate hydrates: current state of the art and future directions, Appl. Energy 122, 112–132. https://doi.org/10.1016/j.apenergy.2014.01.063. [CrossRef] [Google Scholar]
- Mao W.L., Mao H. (2004) Hydrogen storage in molecular compounds, Proceedings of the National Academy of Sciences 101, 3, 708–710. https://doi.org/10.1073/pnas.0307449100 [CrossRef] [PubMed] [Google Scholar]
- Vos W.L., Finger L.W., Hemley R.J., Mao H. (1993) Novel H2–H2O clathrates at high pressures, Phys. Rev. Lett. 71, 19, 3150–3153. https://doi.org/10.1103/PhysRevLett.71.3150. [CrossRef] [PubMed] [Google Scholar]
- Lee H., Lee J., Kim D.Y., Park J., Seo Y.-T., Zeng H., Moudrakovski I.L., Ratcliffe C.I., Ripmeester J.A. (2005) Tuning clathrate hydrates for hydrogen storage, Nature 434, 7034, 743–746. https://doi.org/10.1038/nature03457. [CrossRef] [PubMed] [Google Scholar]
- Feucht K., Hurich W., Komoschinski N., Povei R. (1988) Hydrogen drive for road vehicles-results from the fleet test run in Berlin, Int. J. Hydrogen Energy 13, 4, 243–250. https://doi.org/10.1016/0360-3199(88)90092-4. [CrossRef] [Google Scholar]
- Zaluska A., Zaluski L., Ström-Olsen J.O. (2001) Structure, catalysis and atomic reactions on the nano-scale: a systematic approach to metal hydrides for hydrogen storage, Appl. Phys. A 72, 2, 157–165. https://doi.org/10.1007/s003390100783. [CrossRef] [Google Scholar]
- Dehouche Z., Djaozandry R., Huot J., Boily S., Goyette J., Bose T.K., Schulz R. (2000) Influence of cycling on the thermodynamic and structure properties of nanocrystalline magnesium based hydride, J. Alloys Compd. 305, 1, 264–271. https://doi.org/10.1016/S0925-8388(00)00718-0. [CrossRef] [Google Scholar]
- Bloch J., Mintz M.H. (1997) Kinetics and mechanisms of metal hydrides formation – a review, J. Alloys Compd. 253–254, 529–541. https://doi.org/10.1016/S0925-8388(96)03070-8. [CrossRef] [Google Scholar]
- Reilly J.J., Wiswall R.H. (1974) Formation and properties of iron titanium hydride, Inorg. Chem. 13, 1, 218–222. https://doi.org/10.1021/ic50131a042. [CrossRef] [Google Scholar]
- Sakaguchi H., Tsujimoto T., Adachi G. (1995) The confinement of hydrogen in LaNi5 by poisoning of the hydride surface, J. Alloys Compd. 223, 1, 122–126. https://doi.org/10.1016/0925-8388(94)01489-2. [CrossRef] [MathSciNet] [Google Scholar]
- Myhra S., Kisi E.H., Gray E.M. (1995) A surface analytical study of SO2 stabilisation of LaNi5Hx surfaces, J. Alloys Compd. 224, 2, 305–315. https://doi.org/10.1016/0925-8388(95)01535-3. [CrossRef] [Google Scholar]
- Tarasov B.P., Fursikov P.V., Volodin A.A., Bocharnikov M.S., Shimkus Y.Y., Kashin A.M., Yartys V.A., Chidziva S., Pasupathi S., Lototskyy M.V. (2021) Metal hydride hydrogen storage and compression systems for energy storage technologies, Int. J. Hydrogen Energy 46, 25, 13647–13657. https://doi.org/10.1016/j.ijhydene.2020.07.085. [CrossRef] [Google Scholar]
- Rusman N.A.A., Dahari M. (2016) A review on the current progress of metal hydrides material for solid-state hydrogen storage applications, Int. J. Hydrogen Energy 41, 28, 12108–12126. https://doi.org/10.1016/j.ijhydene.2016.05.244. [CrossRef] [Google Scholar]
- Okada M., Kuriiwa T., Kamegawa A., Takamura H. (2002) Role of intermetallics in hydrogen storage materials, Mater. Sci. Eng. A 329–331, 305–312. https://doi.org/10.1016/S0921-5093(01)01580-5. [CrossRef] [Google Scholar]
- Principi G., Agresti F., Maddalena A., Lo Russo S. (2009) The problem of solid state hydrogen storage, Energy 34, 12, 2087–2091. https://doi.org/10.1016/j.energy.2008.08.027. [CrossRef] [Google Scholar]
- Gasiorowski A., Iwasieczko W., Skoryna D., Drulis H., Jurczyk M. (2004) Hydriding properties of nanocrystalline Mg2−xMxNi alloys synthesized by mechanical alloying (M=Mn, Al), J. Alloys Compd. 364 1, 283–288. https://doi.org/10.1016/S0925-8388(03)00544-9. [CrossRef] [Google Scholar]
- Jain I.P., Lal C., Jain A. (2010) Hydrogen storage in Mg: a most promising material, Int. J. Hydrogen Energy 35, 10, 5133–5144. https://doi.org/10.1016/j.ijhydene.2009.08.088. [CrossRef] [Google Scholar]
- Mushnikov N.V., Ermakov A.E., Uimin M.A., Gaviko V.S., Terent’ev P.B., Skripov A.V., Tankeev A.P., Soloninin A.V., Buzlukov A.L. (2006) Kinetics of interaction of Mg-based mechanically activated alloys with hydrogen, Phys. Metals Metallogr. 102, 4, 421–431. https://doi.org/10.1134/S0031918X06100097. [CrossRef] [Google Scholar]
- Liang G., Huot J., Boily S., Van Neste A., Schulz R. (1999) Catalytic effect of transition metals on hydrogen sorption in nanocrystalline ball milled MgH2–Tm (Tm=Ti, V, Mn, Fe and Ni) systems, J. Alloys Compd. 292, 1, 247–252. https://doi.org/10.1016/S0925-8388(99)00442-9. [CrossRef] [Google Scholar]
- Sadhasivam T., Kim H.-T., Jung S., Roh S.-H., Park J.-H., Jung H.-Y. (2017) Dimensional effects of nanostructured Mg/MgH2 for hydrogen storage applications: a review, Renew. Sust. Energ. Rev. 72, 523–534. https://doi.org/10.1016/j.rser.2017.01.107. [CrossRef] [Google Scholar]
- Shao H., Xin G., Zheng J., Li X., Akiba E. (2012) Nanotechnology in Mg-based materials for hydrogen storage, Nano Energy 1, 4, 590–601. https://doi.org/10.1016/j.nanoen.2012.05.005. [CrossRef] [Google Scholar]
- Fichtner M. (2009) Properties of nanoscale metal hydrides, Nanotechnology 20, 20, 204009. https://doi.org/10.1088/0957-4484/20/20/204009. [CrossRef] [PubMed] [Google Scholar]
- Vajo J.J. (2011) Influence of nano-confinement on the thermodynamics and dehydrogenation kinetics of metal hydrides, Curr. Opin. Solid State Mater. Sci. 15, 2, 52–61. https://doi.org/10.1016/j.cossms.2010.11.001. [CrossRef] [Google Scholar]
- Lai Q., Wang T., Sun Y., Aguey-Zinsou K.-F. (2018) Rational design of nanosized light elements for hydrogen storage: classes, synthesis, characterization, and properties, Adv. Mater. Technol. 3, 9, 1700298. https://doi.org/10.1002/admt.201700298. [CrossRef] [Google Scholar]
- Zhao-Karger Z., Hu J., Roth A., Wang D., Kübel C., Lohstroh W., Fichtner M. (2010) Altered Thermodynamic and Kinetic Properties of MgH2 Infiltrated in Microporous Scaffold, Chem. Commun. 46, 44, 8353–8355. https://doi.org/10.1039/C0CC03072D. [CrossRef] [PubMed] [Google Scholar]
- He T., Cao H., Chen P. (2019) Complex hydrides for energy storage, conversion, and utilization, Adv. Mater. 31, 50, 1902757. https://doi.org/10.1002/adma.201902757. [CrossRef] [Google Scholar]
- Tanaka H., Tokoyoda K., Matsumoto M., Suzuki Y., Kiyobayashi T., Kuriyama N. (2009) Hazard assessment of complex hydrides as hydrogen storage materials, Int. J. Hydrogen Energy 34, 7, 3210–3218. https://doi.org/10.1016/j.ijhydene.2009.01.064. [CrossRef] [Google Scholar]
- Urgnani J., Torres F.J., Palumbo M., Baricco M. (2008) Hydrogen release from solid state NaBH4, Int. J. Hydrogen Energy 33, 12, 3111–3115. https://doi.org/10.1016/j.ijhydene.2008.03.031. [CrossRef] [Google Scholar]
- Bogdanović B., Schwickardi M. (1997) Ti-Doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials, J. Alloys Compd. 253–254, 1–9. https://doi.org/10.1016/S0925-8388(96)03049-6. [CrossRef] [Google Scholar]
- Pohlmann C., Röntzsch L., Hu J., Weißgärber T., Kieback B., Fichtner M. (2012) Tailored heat transfer characteristics of pelletized LiNH2–MgH2 and NaAlH4 hydrogen storage materials, J. Power Sourc. 205, 173–179. https://doi.org/10.1016/j.jpowsour.2012.01.064. [CrossRef] [Google Scholar]
- Züttel A., Wenger P., Rentsch S., Sudan P., Mauron Ph, Emmenegger Ch (2003) LiBH4 a new hydrogen storage material, J. Power Sourc. 118, 1–7. https://doi.org/10.1016/S0378-7753(03)00054-5. [CrossRef] [Google Scholar]
- Chen P., Xiong Z., Luo J., Lin J., Tan K.L. (2002) Interaction of hydrogen with metal nitrides and imides, Nature 420, 6913, 302–304. https://doi.org/10.1038/nature01210. [CrossRef] [PubMed] [Google Scholar]
- Chandra M., Xu Q. (2006) A high-performance hydrogen generation system: transition metal-catalyzed dissociation and hydrolysis of ammonia-borane, J. Power Sourc. 156, 2, 190–194. https://doi.org/10.1016/j.jpowsour.2005.05.043. [CrossRef] [Google Scholar]
- Hua T.Q., Ahluwalia R.K. (2012) Off-board regeneration of ammonia borane for use as a hydrogen carrier for automotive fuel cells, Int. J. Hydrogen Energy 37, 19, 14382–14392. https://doi.org/10.1016/j.ijhydene.2012.07.013. [CrossRef] [Google Scholar]
- Ramachandran P.V., Gagare P.D. (2007) Preparation of ammonia borane in high yield and purity, methanolysis, and regeneration, Inorg. Chem. 46, 19, 7810–7817. https://doi.org/10.1021/ic700772a. [CrossRef] [PubMed] [Google Scholar]
- Al-Kukhun A., Hwang H.T., Varma A. (2013) Mechanistic studies of ammonia borane dehydrogenation, Int. J. Hydrogen Energy 38, 1, 169–179. https://doi.org/10.1016/j.ijhydene.2012.09.161. [CrossRef] [Google Scholar]
- Baitalow F., Baumann J., Wolf G., Jaenicke-Rößler K., Leitner G. (2002) Thermal decomposition of B-N–H compounds investigated by using combined thermoanalytical methods, Thermochim. Acta 391, 1, 159–168. https://doi.org/10.1016/S0040-6031(02)00173-9. [CrossRef] [Google Scholar]
- Frueh S., Kellett R., Mallery C., Molter T., Willis W.S., King’ondu C., Suib S.L. (2011) Pyrolytic decomposition of ammonia borane to boron nitride, Inorg. Chem. 50, 3, 783–792. https://doi.org/10.1021/ic101020k. [CrossRef] [PubMed] [Google Scholar]
- Zhang L., Xia G., Ge Y., Wang C., Guo Z., Li X., Yu X. (2015) Ammonia borane confined by nitrogen-containing carbon nanotubes: enhanced dehydrogenation properties originating from synergetic catalysis and nanoconfinement, J. Mater. Chem. A 3, 41, 20494–20499. https://doi.org/10.1039/C5TA05540G. [CrossRef] [Google Scholar]
- Bluhm M.E., Bradley M.G., Butterick R., Kusari U., Sneddon L.G. (2006) Amineborane-based chemical hydrogen storage: enhanced ammonia borane dehydrogenation in ionic liquids, J. Am. Chem. Soc. 128, 24, 7748–7749. https://doi.org/10.1021/ja062085v. [CrossRef] [PubMed] [Google Scholar]
- Heldebrant D.J., Karkamkar A., Hess N.J., Bowden M., Rassat S., Zheng F., Rappe K., Autrey T. (2008) The effects of chemical additives on the induction phase in solid-state thermal decomposition of ammonia borane, Chem. Mater. 20, 16, 5332–5336. https://doi.org/10.1021/cm801253u. [CrossRef] [Google Scholar]
- Wahab M.A., Zhao H., Yao X.D. (2012) Nano-confined ammonia borane for chemical hydrogen storage, Front. Chem. Sci. Eng. 6, 1, 27–33. https://doi.org/10.1007/s11705-011-1171-3. [CrossRef] [Google Scholar]
- Owarzany R., Jaroń T., Leszczyński P.J., Fijalkowski K.J., Grochala W. (2017) Amidoboranes of rubidium and caesium: the last missing members of the alkali metal amidoborane family, Dalton Trans. 46, 46, 16315–16320. https://doi.org/10.1039/C7DT03590J. [CrossRef] [PubMed] [Google Scholar]
- Ramzan M., Silvearv F., Blomqvist A., Scheicher R.H., Lebègue S., Ahuja R. (2009) Structural and energetic analysis of the hydrogen storage materials LiNH2BH3 and NaNH2BH3 from ab initio calculations, Phys. Rev. B 79, 13, 132102. https://doi.org/10.1103/PhysRevB.79.132102. [CrossRef] [Google Scholar]
- Wu H., Zhou W., Yildirim T. (2008) Alkali and alkaline-earth metal amidoboranes: structure, crystal chemistry, and hydrogen storage properties, J. Am. Chem. Soc. 130, 44, 14834–14839. https://doi.org/10.1021/ja806243f. [CrossRef] [PubMed] [Google Scholar]
- Freepik. Macrovecto, Freepik (accessed 2022-11-30). https://fr.freepik.com/auteur/macrovector [Google Scholar]
- Catalyst: NH3 as an Energy Carrier, Elsevier Enhanced Reader. https://doi.org/10.1016/j.chempr.2017.10.004. [Google Scholar]
- Ammoniac. Techniques de l’Ingénieur (accessed 2022-10-10). https://www.techniques-ingenieur.fr/base-documentaire/procedes-chimie-bio-agro-th2/fabrication-des-grands-produits-industriels-en-chimie-et-petrochimie-42319210/ammoniac-j6135/ [Google Scholar]
- 2014_ifa_ff_ammonia_emissions_july.Pdf (accessed 2022-10-10). https://www.fertilizer.org//images/Library_Downloads/2014_ifa_ff_ammonia_emissions_july.pdf [Google Scholar]
- Rafiqul I., Weber C., Lehmann B., Voss A. (2005) Energy efficiency improvements in ammonia production – perspectives and uncertainties, Energy 30, 13, 2487–2504. https://doi.org/10.1016/j.energy.2004.12.004. [CrossRef] [Google Scholar]
- Wang M., Khan M.A., Mohsin I., Wicks J., Ip A.H., Sumon K.Z., Dinh C.T., Sargent E.H., Gates I.D., Kibria M.G. (2021) Can sustainable ammonia synthesis pathways compete with fossil-fuel based Haber-Bosch processes?, Energy Environ. Sci. 14, 5, 2535–2548. https://doi.org/10.1039/D0EE03808C. [CrossRef] [Google Scholar]
- McEnaney J.M., Singh A.R., Schwalbe J.A., Kibsgaard J., Lin J.C., Cargnello M., Jaramillo T.F., Nørskov J.K. (2017) Ammonia synthesis from N2 and H2O using a lithium cycling electrification strategy at atmospheric pressure, Energy Environ. Sci. 10, 7, 1621–1630. https://doi.org/10.1039/C7EE01126A. [CrossRef] [Google Scholar]
- Kunsman C.H. (1927) The decomposition of ammonia on iron catalysts, Science 65, 1691, 527–528. https://doi.org/10.1126/science.65.1691.527-a. [CrossRef] [PubMed] [Google Scholar]
- Kunsman C.H. (1929) The thermal decomposition of ammonia on iron catalysts II, J. Am. Chem. Soc. 51, 3, 688–695. https://doi.org/10.1021/ja01378a005. [CrossRef] [Google Scholar]
- Lamb K.E., Dolan M.D., Kennedy D.F. (2019) Ammonia for hydrogen storage; a review of catalytic ammonia decomposition and hydrogen separation and purification, Int. J. Hydrogen Energy 44, 7, 3580–3593. https://doi.org/10.1016/j.ijhydene.2018.12.024. [CrossRef] [Google Scholar]
- Makepeace J.W., He T., Weidenthaler C., Jensen T.R., Chang F., Vegge T., Ngene P., Kojima Y., de Jongh P.E., Chen P., David W.I.F. (2019) Reversible ammonia-based and liquid organic hydrogen carriers for high-density hydrogen storage: recent progress, Int. J. Hydrogen Energy 44, 15, 7746–7767. https://doi.org/10.1016/j.ijhydene.2019.01.144. [CrossRef] [Google Scholar]
- Marakatti V.S., Gaigneaux E.M. (2020) Recent advances in heterogeneous catalysis for ammonia synthesis, ChemCatChem 12, 23, 5838–5857. https://doi.org/10.1002/cctc.202001141. [CrossRef] [Google Scholar]
- Chatterjee S., Parsapur R.K., Huang K.-W. (2021) Limitations of ammonia as a hydrogen energy carrier for the transportation sector, ACS Energy Lett. 6, 12, 4390–4394. https://doi.org/10.1021/acsenergylett.1c02189. [CrossRef] [Google Scholar]
- Wang W., Su C., Wu Y., Ran R., Shao Z. (2013) Progress in solid oxide fuel cells with nickel-based anodes operating on methane and related fuels, Chem. Rev. 113, 10, 8104–8151. https://doi.org/10.1021/cr300491e. [CrossRef] [PubMed] [Google Scholar]
- Methanol Industry Installed Capacity and Capital Expenditure (CapEx) Forecast by Region and Countries including details of All Active Plants, Planned and Announced Projects, 2021–2026. Market Research Reports & Consulting | GlobalData UK Ltd. (accessed 2022-10-18). https://www.globaldata.com/store/report/methanol-market-analysis/ [Google Scholar]
- Tian P., Wei Y., Ye M., Liu Z. (2015) Methanol to Olefins (MTO): from fundamentals to commercialization, ACS Catal. 5, 3, 1922–1938. https://doi.org/10.1021/acscatal.5b00007. [CrossRef] [Google Scholar]
- Boocock D.G.B. (March 30, 2004) Process for production of fatty acid methyl esters from fatty acid triglycerides. US6712867B1 (accessed 2022-11-30) https://patents.google.com/patent/US6712867B1/en [Google Scholar]
- Park H., Woo Y., Jung H.S., Kim G., Bae J.W., Park M.-J. (2021) Development of dimethyl ether synthesis processes using by-product gas from a steel-making plant: Single-vs. two-step processes, J. Clean. Prod. 326, 129367. https://doi.org/10.1016/j.jclepro.2021.129367. [CrossRef] [Google Scholar]
- Courty P., Travers C., Durand D., Forestière A., Chaumette P. (1987) Process for the preparation of catalysts comprising copper, zinc and aluminium, useful in the production of methanol from synthesis gas, EP0152314B1 August 12 (accessed 2022-11-30) https://patents.google.com/patent/EP0152314B1/en [Google Scholar]
- Magoon E. (1973) Production of Methanol. US3709919A January 9, 1973 (accessed 2022-11-30) https://patents.google.com/patent/US3709919A/en?oq=+Shell%2c+US+3709919%2c+1973+(E.F.+Magoon) [Google Scholar]
- Gallagher J.T., Kidd J.M. (1969) Methanol Synthesis. GB1159035A, July 23, 1969 (accessed 2022-11-30) https://patents.google.com/patent/GB1159035A/en?oq=GB+1159035 [Google Scholar]
- Schneider M.D.D.-C., Kochloefl K.D.D.-C., Ladebeck J.D.D.-C. (June 16, 1987.) Katalysator Für Die Methanolsynthese. EP0125689B1 June 16, 1987 (accessed 2022-11-30) https://patents.google.com/patent/EP0125689B1/en?oq=EP+0125689 [Google Scholar]
- Höppener R.H., Doesburg E.B.M., Scholten J.J.F. (1986) Preparation and characterization of stable copper/zinc oxide/alumina catalysts for methanol systhesis, Appl. Catal. 25, 1, 109–119. https://doi.org/10.1016/S0166-9834(00)81227-0. [CrossRef] [Google Scholar]
- Shamsul N.S., Kamarudin S.K., Rahman N.A., Kofli N.T. (2014) An overview on the production of bio-methanol as potential renewable energy, Renew. Sust. Energ. Rev. 33, 578–588. https://doi.org/10.1016/j.rser.2014.02.024. [CrossRef] [Google Scholar]
- Xie S., Zhang W., Lan X., Lin H. (2020) CO2 reduction to methanol in the liquid phase: a review, ChemSusChem 13, 23, 6141–6159. https://doi.org/10.1002/cssc.202002087. [CrossRef] [PubMed] [Google Scholar]
- Huff C.A., Sanford M.S. (2011) Cascade catalysis for the homogeneous hydrogenation of CO2 to methanol, J. Am. Chem. Soc. 133, 45, 18122–18125. https://doi.org/10.1021/ja208760j. [CrossRef] [PubMed] [Google Scholar]
- Miller A.J.M., Heinekey D.M., Mayer J.M., Goldberg K.I. (2013) Catalytic disproportionation of formic acid to generate methanol, Angew. Chem. Int. Ed. Engl. 52, 14, 3981–3984. https://doi.org/10.1002/anie.201208470. [CrossRef] [PubMed] [Google Scholar]
- Kanega R., Onishi N., Tanaka S., Kishimoto H., Himeda Y. (2021) Catalytic hydrogenation of CO2 to methanol using multinuclear iridium complexes in a gas-solid phase reaction, J. Am. Chem. Soc. 143, 3, 1570–1576. https://doi.org/10.1021/jacs.0c11927. [CrossRef] [PubMed] [Google Scholar]
- Sá S., Silva H., Brandão L., Sousa J.M., Mendes A. (2010) Catalysts for methanol steam reforming – a review, Appl. Catal. B Environ. 99, 1, 43–57. https://doi.org/10.1016/j.apcatb.2010.06.015. [CrossRef] [Google Scholar]
- Eberle U., Felderhoff M., Schüth F. (2009) Chemical and physical solutions for hydrogen storage, Angew. Chem. Int. Ed. 48, 36, 6608–6630. https://doi.org/10.1002/anie.200806293. [CrossRef] [PubMed] [Google Scholar]
- Hodoshima S., Arai H., Takaiwa S., Saito Y. (2003) Catalytic decalin dehydrogenation/naphthalene hydrogenation pair as a hydrogen source for fuel-cell vehicle, Int. J. Hydrogen Energy 28, 11, 1255–1262. https://doi.org/10.1016/S0360-3199(02)00250-1. [CrossRef] [Google Scholar]
- Sultan O., Shaw H. (1975) Study of automotive storage of hydrogen using recyclable liquid chemical carriers [Catalytic Dehydrogenation of Naphthenes]; TEC-75/003, Government Research Lab., Exxon Research and Engineering Co., Linden, NJ, USA (accessed 2020-03-03). https://www.osti.gov/biblio/5000657 [Google Scholar]
- Taube M., Taube P. (1979) A liquid organic carrier of hydrogen as a fuel for automobiles; EIR–379, Eidgenoessisches Inst. fuer Reaktorforschung (accessed 2020-03-03) http://inis.iaea.org/Search/search.aspx?orig_q=RN:13652276 [Google Scholar]
- Grünenfelder N.F., Schucan ThH (1989) Seasonal storage of hydrogen in liquid organic hydrides: description of the second prototype vehicle, Int. J. Hydrogen Energy 14, 8, 579–586. https://doi.org/10.1016/0360-3199(89)90117-1. [CrossRef] [Google Scholar]
- He T., Pei Q., Chen P. (2015) Liquid organic hydrogen carriers, J. Energy Chem. 24, 5, 587–594. https://doi.org/10.1016/j.jechem.2015.08.007. [CrossRef] [Google Scholar]
- Züttel A. (2004) Hydrogen storage methods, Naturwissenschaften 91, 4, 157–172. https://doi.org/10.1007/s00114-004-0516-x. [CrossRef] [PubMed] [Google Scholar]
- Riha M. (2020) Hydrogenious LOHC Technologies GmbH, FUTURIUM – European Commission. https://ec.europa.eu/futurium/en/tech-society-2020/hydrogenious-lohc-technologies-gmbh (accessed 2022-11-02). [Google Scholar]
- Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles. Energy.gov (accessed 2021-06-04). https://www.energy.gov/eere/fuelcells/downloads/target-explanation-document-onboard-hydrogen-storage-light-duty-fuel-cell [Google Scholar]
- Geburtig D., Preuster P., Bösmann A., Müller K., Wasserscheid P. (2016) Chemical utilization of hydrogen from fluctuating energy sources – catalytic transfer hydrogenation from charged liquid organic hydrogen carrier systems, Int. J. Hydrogen Energy 41, 2, 1010–1017. https://doi.org/10.1016/j.ijhydene.2015.10.013. [CrossRef] [Google Scholar]
- Teichmann D., Arlt W., Wasserscheid P. (2012) Liquid organic hydrogen carriers as an efficient vector for the transport and storage of renewable energy, Int. J. Hydrogen Energy 37, 23, 18118–18132. https://doi.org/10.1016/j.ijhydene.2012.08.066. [CrossRef] [Google Scholar]
- Aakko-Saksa P.T., Cook C., Kiviaho J., Repo T. (2018) Liquid organic hydrogen carriers for transportation and storing of renewable energy – review and discussion, J. Power Sourc. 396, 803–823. https://doi.org/10.1016/j.jpowsour.2018.04.011. [CrossRef] [Google Scholar]
- Niermann M., Beckendorff A., Kaltschmitt M., Bonhoff K. (2019) Liquid Organic Hydrogen Carrier (LOHC) – assessment based on chemical and economic properties, Int. J. Hydrogen Energy 44, 13, 6631–6654. https://doi.org/10.1016/j.ijhydene.2019.01.199. [CrossRef] [Google Scholar]
- Modisha P.M., Ouma C.N.M., Garidzirai R., Wasserscheid P., Bessarabov D. (2019) The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers, Energy Fuels 33, 4, 2778–2796. https://doi.org/10.1021/acs.energyfuels.9b00296. [CrossRef] [Google Scholar]
- Rao P.C., Yoon M. (2020) Potential Liquid-Organic Hydrogen Carrier (LOHC) systems: a review on recent progress, Energies 13, 22, 6040. https://doi.org/10.3390/en13226040. [CrossRef] [Google Scholar]
- Cho J.-Y., Kim H., Oh J.-E., Park B.Y. (2021) Recent advances in homogeneous/heterogeneous catalytic hydrogenation and dehydrogenation for potential Liquid Organic Hydrogen Carrier (LOHC) systems, Catalysts 11, 12, 1497. https://doi.org/10.3390/catal11121497. [CrossRef] [Google Scholar]
- Zelinsky N. (1911) Über Dehydrogenisation Durch Katalyse, Berichte der deutschen chemischen Gesellschaft 44, 3, 3121–3125. https://doi.org/10.1002/cber.191104403168. [CrossRef] [Google Scholar]
- Zelinsky N. (1912) Über Die Selektive Dehydrogenisations-Katalyse, Berichte der deutschen chemischen Gesellschaft 45, 3, 3678–3682. https://doi.org/10.1002/cber.191204503126. [CrossRef] [Google Scholar]
- Cooper A.C. (2012) Design and development of new carbon-based sorbent systems for an effective containment of hydrogen, Air Products and Chemicals Inc., pp. 86–377-P, 1039432. https://doi.org/10.2172/1039432 [Google Scholar]
- Clar E. (1983) The aromatic sextet, in Mobile source emissions including policyclic organic species, D. Rondia, M. Cooke, R.K. Haroz (eds.), NATO ASI Series. Springer, Dordrecht, Netherlands, pp. 49–58. https://doi.org/10.1007/978-94-009-7197-4_4 [CrossRef] [Google Scholar]
- Solà M. (2013) Forty years of Clar’s aromatic π-sextet rule, Front. Chem. 1. https://doi.org/10.3389/fchem.2013.00022 [Google Scholar]
- Pumera M., An Wong C.H. (2013) Graphane and hydrogenated graphene, Chem. Soc.Rev. 42, 14, 5987–5995. https://doi.org/10.1039/C3CS60132C. [CrossRef] [PubMed] [Google Scholar]
- Luo Z., Yu T., Kim K., Ni Z., You Y., Lim S., Shen Z., Wang S., Lin J. (2009) Thickness-dependent reversible hydrogenation of graphene layers, ACS Nano. 3, 7, 1781–1788. https://doi.org/10.1021/nn900371t. [CrossRef] [PubMed] [Google Scholar]
- Kalenchuk A., Bogdan V., Dunaev S., Kustov L. (2020) Influence of steric factors on reversible reactions of hydrogenation-dehydrogenation of polycyclic aromatic hydrocarbons on a Pt/C catalyst in hydrogen storage systems, Fuel 280, 118625. https://doi.org/10.1016/j.fuel.2020.118625. [CrossRef] [Google Scholar]
- Hydrogen, C. S. Mitsubishi Hitachi Power Systems Ltd. Development Bank of Japan Inc. 3 [Google Scholar]
- Vanhanen J.P., Lund P.D. (1995) Computational approaches for improving seasonal storage systems based on hydrogen technologies, Int. J. Hydrogen Energy 20, 7, 575–585. https://doi.org/10.1016/0360-3199(94)00110-L. [CrossRef] [Google Scholar]
- Taube M., Rippin D., Knecht W., Hakimifard D., Milisavljevic B., Gruenenfelder N. (1985) A prototype truck powered by hydrogen from organic liquid hydrides, Int. J. Hydrogen Energy 10, 9, 595–599. https://doi.org/10.1016/0360-3199(85)90035-7. [CrossRef] [Google Scholar]
- Kerleau P., Swesi Y., Meille V., Pitault I., Heurtaux F. (2010) Total catalytic oxidation of a side-product for an autothermal restoring hydrogen process, Catal. Today 157, 1, 321–326. https://doi.org/10.1016/j.cattod.2010.02.011. [CrossRef] [Google Scholar]
- Touzani A., Klvana D., Bélanger G. (1984) Dehydrogenation reactor for a vehicle equipped with a hydrogen engine: a simulation study, Int. J. Hydrogen Energy 9, 11, 929–936. https://doi.org/10.1016/0360-3199(84)90158-7. [CrossRef] [Google Scholar]
- Niimi T., Nagasawa H., Kanezashi M., Yoshioka T., Ito K., Tsuru T. (2014) Preparation of BTESE-derived organosilica membranes for catalytic membrane reactors of methylcyclohexane dehydrogenation, J. Membrane Sci. 455, 375–383. https://doi.org/10.1016/j.memsci.2014.01.003. [CrossRef] [Google Scholar]
- Ali J.K., Newson E.J., Rippin D.W.T. (1994) Exceeding equilibrium conversion with a catalytic membrane reactor for the dehydrogenation of methylcyclohexane, Chem. Eng. Sci. 49, 13, 2129–2134. https://doi.org/10.1016/0009-2509(94)E0035-O. [CrossRef] [Google Scholar]
- Pal N., Agarwal M., Maheshwari K., Solanki Y.S. (2020) A review on types, fabrication and support material of hydrogen separation membrane, Mater. Today Proc. 28, 1386–1391. https://doi.org/10.1016/j.matpr.2020.04.806. [CrossRef] [Google Scholar]
- Chytil S., Glomm W.R., Vollebekk E., Bergem H., Walmsley J., Sjöblom J., Blekkan E.A. (2005) Platinum nanoparticles encapsulated in mesoporous silica: preparation, characterisation and catalytic activity in toluene hydrogenation, Microporous Mesoporous Mater. 86, 1, 198–206. https://doi.org/10.1016/j.micromeso.2005.06.037. [CrossRef] [Google Scholar]
- Alexeev O., Gates B.C. (1998) Iridium clusters supported on γ-Al2O3: structural characterization and catalysis of toluene hydrogenation, J. Catal. 176, 2, 310–320. https://doi.org/10.1006/jcat.1998.2053. [CrossRef] [Google Scholar]
- Weber W.A., Gates B.C. (1998) Rhodium supported on faujasites: effects of cluster size and CO ligands on catalytic activity for toluene hydrogenation, J. Catal. 180, 2, 207–217. https://doi.org/10.1006/jcat.1998.2264. [CrossRef] [Google Scholar]
- Suppino R.S., Landers R., Cobo A.J.G. (2016) Influence of Noble metals (Pd, Pt) on the performance of Ru/Al2O3 based catalysts for toluene hydrogenation in liquid phase, Appl. Catal. A Gen. 525, 41–49. https://doi.org/10.1016/j.apcata.2016.06.038. [CrossRef] [Google Scholar]
- Rousset J.L., Stievano L., Aires F.J.C.S., Geantet C., Renouprez A.J., Pellarin M. (2001) Hydrogenation of toluene over γ-Al2O3-Supported Pt, Pd, and Pd–Pt model catalysts obtained by laser vaporization of bulk metals, J. Catal. 197, 2, 335–343. https://doi.org/10.1006/jcat.2000.3083. [CrossRef] [Google Scholar]
- Thomas K., Binet C., Chevreau T., Cornet D., Gilson J.-P. (2002) Hydrogenation of toluene over supported Pt and Pd catalysts: influence of structural factors on the sulfur tolerance, J. Catal. 212, 1, 63–75. https://doi.org/10.1006/jcat.2002.3780. [CrossRef] [Google Scholar]
- Lindfors L.P., Salmi T., Smeds S. (1993) Kinetics of toluene hydrogenation on Ni/Al2O3 catalyst, Chem. Eng. Sci. 48, 22, 3813–3828. https://doi.org/10.1016/0009-2509(93)80224-E. [CrossRef] [Google Scholar]
- Shuwa S.M., Jibril B.Y., Al-Hajri R.S. (2017) Hydrogenation of toluene on Ni-Co-Mo supported zeolite catalysts, Nigerian J. Technol. 36, 4, 1114–1123. https://doi.org/10.4314/njt.v36i4.17. [Google Scholar]
- Wang J., Huang L., Li Q. (1998) Influence of different diluents in Pt/Al2O3 catalyst on the hydrogenation of benzene, toluene and o-xylene, Appl. Catal. A Gen. 175, 1, 191–199. https://doi.org/10.1016/S0926-860X(98)00216-6. [CrossRef] [Google Scholar]
- Zhu L., Zhang H., Hu W., Zheng J., Zhang N., Yu C., Ye H., Yang Z., Chen B.H. (2018) Nickel Hydroxide-Cobalt Hydroxide Nanoparticle Supported Ruthenium–Nickel–Cobalt Islands as an Efficient Nanocatalyst for the Hydrogenation Reaction, ChemCatChem 10, 9, 1998–2002. https://doi.org/10.1002/cctc.201701847. [CrossRef] [Google Scholar]
- Okada Y., Sasaki E., Watanabe E., Hyodo S., Nishijima H. (2006) Development of dehydrogenation catalyst for hydrogen generation in organic chemical hydride method, Int. J. Hydrogen Energy 31, 10, 1348–1356. https://doi.org/10.1016/j.ijhydene.2005.11.014. [CrossRef] [Google Scholar]
- Wang J., Liu H., Fan S., Wang S., Xu G., Guo A., Wang Z. (2021) Dehydrogenation of cycloalkanes over N-doped carbon-supported catalysts: the effects of active component and molecular structure of the substrate, Nanomaterials 11, 11, 2846. https://doi.org/10.3390/nano11112846. [CrossRef] [PubMed] [Google Scholar]
- Shukla A.A., Gosavi P.V., Pande J.V., Kumar V.P., Chary K.V.R., Biniwale R.B. (2010) Efficient hydrogen supply through catalytic dehydrogenation of methylcyclohexane over Pt/metal oxide catalysts, Int. J. Hydrogen Energy 35, 9, 4020–4026. https://doi.org/10.1016/j.ijhydene.2010.02.014. [CrossRef] [Google Scholar]
- Wu X., Lu H., Xiao Y., Guo H., Jia L., Li D. (2022) Acid site introduced by Al3+ penta and boron in Pt/Al2O3 catalyst for dehydrogenation of methylcyclohexane, Int. J. Hydrogen Energy 47, 82. https://doi.org/10.1016/j.ijhydene.2022.08.085. [Google Scholar]
- Parra C.F., Goldwasser M.R., Fajula F., Figueras F. (1985) A study of the hydrogen transfer reaction between isobutene and cyclohexane over zeolites using carbon-13 labelled molecules, Appl. Catal. 17, 2, 217–222. https://doi.org/10.1016/S0166-9834(00)83206-6. [CrossRef] [Google Scholar]
- de la Banda J.F.G., Melo A.C.Y.F.V. (1986) Dehydrogenation of methylcyclohexene on a PtNaY catalyst. Study of kinetics and deactivation, Appl. Catal. 26, 103–121. https://doi.org/10.1016/S0166-9834(00)82545-2. [CrossRef] [Google Scholar]
- Van Trimpont P.A., Marin G.B., Froment G.F. (1986) Kinetics of methylcyclohexane dehydrogenation on sulfided commercial platinum/alumina and platinum-rhenium/alumina catalysts, Ind. Eng. Chem. Fund. 25, 4, 544–553. https://doi.org/10.1021/i100024a014. [CrossRef] [Google Scholar]
- Chen L., Verma P., Hou K., Qi Z., Zhang S., Liu Y.-S., Guo J., Stavila V., Allendorf M.D., Zheng L., Salmeron M., Prendergast D., Somorjai G.A., Su J. (2022) Reversible dehydrogenation and rehydrogenation of cyclohexane and methylcyclohexane by single-site platinum catalyst, Nat. Commun. 13, 1, 1092. https://doi.org/10.1038/s41467-022-28607-y. [CrossRef] [MathSciNet] [Google Scholar]
- Sebastian O., Nair S., Taccardi N., Wolf M., Søgaard A., Haumann M., Wasserscheid P. (2020) Stable and selective dehydrogenation of methylcyclohexane using supported catalytically active liquid metal solutions – Ga52Pt/SiO2 SCALMS, ChemCatChem 12, 18, 4533–4537. https://doi.org/10.1002/cctc.202000671. [CrossRef] [Google Scholar]
- Manabe R., Okada S., Inagaki R., Oshima K., Ogo S., Sekine Y. (2016) Surface protonics promotes catalysis, Sci. Rep. 6, 1, 38007. https://doi.org/10.1038/srep38007. [CrossRef] [Google Scholar]
- Kosaka M., Higo T., Ogo S., Seo J.G., Kado S., Imagawa K., Sekine Y. (2020) Low-temperature selective dehydrogenation of methylcyclohexane by surface protonics over Pt/Anatase-TiO2 catalyst, Int. J. Hydrogen Energy 45, 1, 738–743. https://doi.org/10.1016/j.ijhydene.2019.10.133. [CrossRef] [Google Scholar]
- Yolcular S., Olgun Ö. (2008) Ni/Al2O3 catalysts and their activity in dehydrogenation of methylcyclohexane for hydrogen production, Catal. Today 138, 3, 198–202. https://doi.org/10.1016/j.cattod.2008.07.020. [CrossRef] [Google Scholar]
- Al-ShaikhAli H., Jedidi A., Cavallo L., Takanabe K. (2015) Non-precious bimetallic catalysts for selective dehydrogenation of an organic chemical hydride system, Chem. Commun. 51 65, 12931–12934. https://doi.org/10.1039/C5CC04016G. [CrossRef] [PubMed] [Google Scholar]
- Al-ShaikhAli A.H., Jedidi A., Anjum D.H., Cavallo L., Takanabe K. (2017) Kinetics on NiZn bimetallic catalysts for hydrogen evolution via selective dehydrogenation of methylcyclohexane to toluene, ACS Catal. 7, 3, 1592–1600. https://doi.org/10.1021/acscatal.6b03299. [CrossRef] [Google Scholar]
- Zhao W., Chizallet C., Sautet P., Raybaud P. (2019) Dehydrogenation mechanisms of methyl-cyclohexane on γ-Al2O3 Supported Pt13: impact of cluster ductility, J. Catal. 370, 118–129. https://doi.org/10.1016/j.jcat.2018.12.004. [CrossRef] [Google Scholar]
- Zhao, W. Dehydrogenation mechanisms of methyl-cyclohexane on γ-alumina supported platinum subnanometric-clusters : DFT coupled with experimental kinetics and kinetic modellingdocument.pdf. HAL archives-ouvertes.fr. (accessed 2020-06-29). https://tel.archives-ouvertes.fr/tel-01827240/document. [Google Scholar]
- Obodo K.O., Ouma C.N.M., Bessarabov D. (2022) Low-Pt-based Sn alloy for the dehydrogenation of methylcyclohexane to toluene: a density functional theory study, Catalysts 12, 10, 1221. https://doi.org/10.3390/catal12101221. [CrossRef] [Google Scholar]
- Auer F., Blaumeiser D., Bauer T., Bösmann A., Szesni N., Libuda J., Wasserscheid P. (2019) Boosting the activity of hydrogen release from liquid organic hydrogen carrier systems by sulfur-additives to Pt on alumina catalysts, Catal. Sci. Technol. 9, 13, 3537–3547. https://doi.org/10.1039/C9CY00817A. [CrossRef] [Google Scholar]
- Ouma C.N.M., Obodo K.O., Modisha P.M., Bessarabov D. (2022) Si, P, S and Se surface additives as catalytic activity boosters for dehydrogenation of methylcyclohexane to toluene – a liquid organic hydrogen carrier system: density functional theory insights, Mater. Chem. Phys. 2022, 125728. https://doi.org/10.1016/j.matchemphys.2022.125728 [CrossRef] [Google Scholar]
- Zhang C., Song P., Zhang Y., Xiao L., Hou J., Wang X. (2022) Technical and cost analysis of imported hydrogen based on MCH-TOL hydrogen storage technology, Int. J. Hydrog. Energy. 47, 27717–27732. https://doi.org/10.1016/j.ijhydene.2022.06.113. [CrossRef] [Google Scholar]
- Informatics, N. O. of D.. WebBook de Chimie NIST. https://doi.org/10.18434/T4D303 [Google Scholar]
- Popescu A.I., Bombos M., Doukeh R., Bombos D., Bolocan I. (2016) Acidity influence of Ru catalysts on the hydrogenation of naphtalene, Rev. Chim. 67, 3, 5. [Google Scholar]
- Popescu (Stanica) A.I., Bombos M., Popovici R.D., Bombos D., Bolocan I. (2017) Hydrogenation of Naphtalene on Pt-Pd Catalyst, Rev. Chim. 68 2, 210–214. https://doi.org/10.37358/RC.17.2.5421 [CrossRef] [Google Scholar]
- Park K.-C., Yim D.-J., Ihm S.-K. (2002) Characteristics of Al-MCM-41 supported Pt catalysts: effect of Al distribution in Al-MCM-41 on its catalytic activity in naphthalene hydrogenation, Catal. Today 74, 3, 281–290. https://doi.org/10.1016/S0920-5861(02)00024-X. [CrossRef] [Google Scholar]
- Manríquez M.E., Hernandez-Pichardo M.L., Barrera M.C., Ramírez-López R., Castro L.V. (2018) Enhanced catalytic activity on the naphtalene hydrogenation reaction over Pt-Pd/Al2O3-CeO2 catalysts, Rev. Mex. Ing. Quím. 17, 3, 913–925. https://doi.org/10.24275/uam/izt/dcbi/revmexingquim/2018v17n3/Manriquez [CrossRef] [Google Scholar]
- Montesinos-Castellanos A., Zepeda T.A. (2008) High hydrogenation performance of the mesoporous NiMo/Al(Ti, Zr)–HMS catalysts, Microporous Mesoporous Mater. 113, 1, 146–162. https://doi.org/10.1016/j.micromeso.2007.11.012. [CrossRef] [Google Scholar]
- Kariya N., Fukuoka A., Ichikawa M. (2002) Efficient evolution of hydrogen from liquid cycloalkanes over pt-containing catalysts supported on active carbons under “Wet–Dry Multiphase Conditions”, Appl. Catal. A Gen. 233, 1, 91–102. https://doi.org/10.1016/S0926-860X(02)00139-4. [CrossRef] [Google Scholar]
- Shinohara C., Kawakami S., Moriga T., Hayashi H., Hodoshima S., Saito Y., Sugiyama S. (2004) Local structure around platinum in Pt/C Catalysts employed for liquid-phase dehydrogenation of decalin in the liquid-film state under reactive distillation conditions, Appl. Catal. A Gen. 266, 2, 251–255. https://doi.org/10.1016/j.apcata.2004.02.014. [CrossRef] [Google Scholar]
- Kariya N., Fukuoka A., Utagawa T., Sakuramoto M., Goto Y., Ichikawa M. (2003) Efficient hydrogen production using cyclohexane and decalin by pulse-spray mode reactor with Pt catalysts, Appl. Catal. A Gen. 247, 2, 247–259. https://doi.org/10.1016/S0926-860X(03)00104-2. [CrossRef] [Google Scholar]
- Ninomiya W., Tanabe Y., Sotowa K.-I., Yasukawa T., Sugiyama S. (2008) Dehydrogenation of cycloalkanes over noble metal catalysts supported on active carbon, Res. Chem. Intermed. 34, 8, 663–668. https://doi.org/10.1007/BF03036923. [CrossRef] [Google Scholar]
- Kim K., Oh J., Kim T.W., Park J.H., Han J.W., Suh Y.-W. (2017) Different catalytic behaviors of Pd and Pt metals in decalin dehydrogenation to naphthalene, Catal. Sci. Technol. 7, 17, 3728–3735. https://doi.org/10.1039/C7CY00569E. [CrossRef] [Google Scholar]
- Wang B., Goodman D.W., Froment G.F. (2008) Kinetic modeling of pure hydrogen production from decalin, J. Catal. 253, 2, 229–238. https://doi.org/10.1016/j.jcat.2007.11.012. [CrossRef] [Google Scholar]
- Martynenko E.A., Pimerzin Al.A., Savinov A.A., Verevkin S.P., Pimerzin A.A. (2020) Hydrogen release from decalin by catalytic dehydrogenation over supported platinum catalysts, Top. Catal. 63, 1, 178–186. https://doi.org/10.1007/s11244-020-01228-9. [CrossRef] [Google Scholar]
- Lee G., Jeong Y., Kim B.-G., Han J.S., Jeong H., Na H.B., Jung J.C. (2015) Hydrogen production by catalytic decalin dehydrogenation over carbon-supported platinum catalyst: effect of catalyst preparation method, Catal. Commun. 67, 40–44. https://doi.org/10.1016/j.catcom.2015.04.002. [CrossRef] [Google Scholar]
- Wang Z., Liu G., Zhang X. (2023) Efficient and stable Pt/CaO-TiO2-Al2O3 for the catalytic dehydrogenation of cycloalkanes as an endothermic hydrocarbon fuel, Fuel 331, 125732. https://doi.org/10.1016/j.fuel.2022.125732. [CrossRef] [Google Scholar]
- Jiang N., Rao K.S.R., Jin M.-J., Park S.-E. (2012) Effect of hydrogen spillover in decalin dehydrogenation over supported Pt catalysts, Appl. Catal. A Gen. 425–426, 62–67. https://doi.org/10.1016/j.apcata.2012.03.001. [CrossRef] [Google Scholar]
- Li P., Huang Y.-L., Chen D., Zhu J., Zhao T.-J., Zhou X.-G. (2009) CNFs-supported Pt catalyst for hydrogen evolution from decalin, Catal. Commun. 10, 6, 815–818. https://doi.org/10.1016/j.catcom.2008.12.004. [CrossRef] [Google Scholar]
- Tuo Y.-X., Shi L.-J., Cheng H.-Y., Zhu Y.-A., Yang M.-L., Xu J., Han Y.-F., Li P., Yuan W.-K. (2018) Insight into the support effect on the particle size effect of Pt/C catalysts in dehydrogenation, J Catal. 360, 175–186. https://doi.org/10.1016/j.jcat.2018.02.001. [CrossRef] [Google Scholar]
- Tuo Y., Yang L., Ma X., Ma Z., Gong S., Li P. (2021) Carbon nanotubes-supported Pt catalysts for decalin dehydrogenation to release hydrogen: a comparison between nitrogen- and oxygen-surface modification, Int. J. Hydrogen Energy 46, 1, 930–942. https://doi.org/10.1016/j.ijhydene.2020.09.225. [CrossRef] [Google Scholar]
- Tuo Y., Meng Y., Chen C., Lin D., Feng X., Pan Y., Li P., Chen D., Liu Z., Zhou Y., Zhang J. (2021) Partial positively charged Pt in Pt/MgAl2O4 for enhanced dehydrogenation activity, Appl. Catal. B: Environ. 288, 119996. https://doi.org/10.1016/j.apcatb.2021.119996. [CrossRef] [Google Scholar]
- Tuo Y., Yang L., Cheng H., Yang M., Zhu Y.-A., Li P. (2018) Density functional theory study of decalin dehydrogenation for hydrogen release on Pt(111) and Pt(211), Int. J. Hydrogen Energy 43, 42, 19575–19588. https://doi.org/10.1016/j.ijhydene.2018.09.002. [CrossRef] [Google Scholar]
- Wang Y., Shah N., Huggins F.E., Huffman G.P. (2006) Hydrogen production by catalytic dehydrogenation of tetralin and decalin over stacked cone carbon nanotube-supported Pt catalysts, Energy Fuels 20, 6, 2612–2615. https://doi.org/10.1021/ef060228t. [CrossRef] [Google Scholar]
- Kalenchuk A.N., Smetneva D.N., Bogdan V.I., Kustov L.M. (2015) Kinetics of decalin dehydrogenation on Pt/C catalyst, Russ. Chem. Bull. 64, 11, 2642–2645. https://doi.org/10.1007/s11172-015-1202-1. [CrossRef] [Google Scholar]
- Martynenko E.A., Vostrikov S.V., Pimerzin A.A. (2021) Hydrogen production from decalin over silica-supported platinum catalysts: a kinetic and thermodynamic study, Reac. Kinet. Mech. Cat. 133, 2, 713–728. https://doi.org/10.1007/s11144-021-02037-1. [CrossRef] [Google Scholar]
- Dziewiecki Z., Ihnatowicz M., Makowski A. (1979) Activity of Ni-Moly catalysts in tetralin or decalin dehydrogenation and in hydrogenation of coal-extract solution, Fuel 58, 10, 737–740. https://doi.org/10.1016/0016-2361(79)90072-3. [CrossRef] [Google Scholar]
- Qi S., Li Y., Yue J., Chen H., Yi C., Yang B. (2014) Hydrogen production from decalin dehydrogenation over Pt-Ni/C bimetallic catalysts, Chin. J. Catal. 35, 11, 1833–1839. https://doi.org/10.1016/S1872-2067(14)60178-9. [CrossRef] [Google Scholar]
- Patil S.P., Pande J.V., Biniwale R.B. (2013) Non-noble Ni–Cu/ACC bimetallic catalyst for dehydrogenation of liquid organic hydrides for hydrogen storage, Int. J. Hydrogen Energy 38, 35, 15233–15241. https://doi.org/10.1016/j.ijhydene.2013.09.115. [CrossRef] [Google Scholar]
- Qi S., Yue J., Li Y., Huang J., Yi C., Yang B. (2014) Replacing platinum with tungsten carbide for decalin dehydrogenation, Catal. Lett. 144, 8, 1443–1449. https://doi.org/10.1007/s10562-014-1284-7. [CrossRef] [Google Scholar]
- Brückner N., Obesser K., Bösmann A., Teichmann D., Arlt W., Dungs J., Wasserscheid P. (2014) Evaluation of industrially applied heat-transfer fluids as liquid organic hydrogen carrier systems, ChemSusChem 7, 1, 229–235. https://doi.org/10.1002/cssc.201300426. [CrossRef] [PubMed] [Google Scholar]
- Müller K., Stark K., Emely’anenko V.N., Varfolomeev M.A., Zaitsau D.H., Shoifet E., Schick C., Verevkin S.P., Arlt W. (2015) Liquid organic hydrogen carriers: thermophysical and thermochemical studies of benzyl- and dibenzyl-toluene derivatives, Ind. Eng. Chem. Res. 54, 32, 7967–7976. https://doi.org/10.1021/acs.iecr.5b01840. [CrossRef] [Google Scholar]
- Marlotherm-SH-SDS.Pdf. (accessed 2022-11-10). https://chem-group.com/wp-content/uploads/2020/07/Marlotherm-SH-SDS.pdf [Google Scholar]
- 0_HydrogeniousTechnologies.Pdf (accessed 2022-11-09). https://www.energiewende-erlangen.de/wp-content/uploads/2018/02/0_HydrogeniousTechnologies.pdf [Google Scholar]
- Kim T.W., Jeong H., Jo Y., Kim D., Park J.H., Kim S.K., Suh Y.-W. (2022) Advanced heterolytic H2 adsorption of K-added Ru/MgO catalysts for accelerating hydrogen storage into aromatic benzyltoluenes, J. Energy Chem., 71, 333–343. https://doi.org/10.1016/j.jechem.2022.03.047. [CrossRef] [Google Scholar]
- Do G., Preuster P., Aslam R., Bösmann A., Müller K., Arlt W., Wasserscheid P. (2016) Hydrogenation of the liquid organic hydrogen carrier compound dibenzyltoluene – reaction pathway determination by 1 H NMR spectroscopy, Reac. Chem. Eng. 1, 3, 313–320. https://doi.org/10.1039/C5RE00080G. [CrossRef] [Google Scholar]
- Jorschick H., Preuster P., Dürr S., Seidel A., Müller K., Bösmann A., Wasserscheid P. (2017) Hydrogen storage using a hot pressure swing reactor, Energy Environ. Sci. 10, 7, 1652–1659. https://doi.org/10.1039/C7EE00476A. [CrossRef] [Google Scholar]
- Shi L., Qi S., Qu J., Che T., Yi C., Yang B. (2019) Integration of hydrogenation and dehydrogenation based on dibenzyltoluene as liquid organic hydrogen energy carrier, Int. J. Hydrogen Energy 44, 11, 5345–5354. https://doi.org/10.1016/j.ijhydene.2018.09.083. [CrossRef] [Google Scholar]
- Ali A., Rohini A.K., Noh Y.S., Moon D.J., Lee H.J. (2022) Hydrogenation of dibenzyltoluene and the catalytic performance of Pt/Al2O3 with various Pt loadings for hydrogen production from perhydro-dibenzyltoluene, Inter. J. Energy Res. 46, 5, 6672–6688. https://doi.org/10.1002/er.7604. [CrossRef] [Google Scholar]
- Modisha P., Bessarabov D. (2020) Stress tolerance assessment of dibenzyltoluene-based liquid organic hydrogen carriers, Sustain. Energy Fuels 4, 9, 4662–4670. https://doi.org/10.1039/D0SE00625D. [CrossRef] [Google Scholar]
- Feng X., Jiang L., Li Z., Wang S., Ye J., Wu Y., Yuan B. (2022) Boosting the hydrogenation activity of dibenzyltoluene catalyzed by Mg-based metal hydrides, Int. J. Hydrogen Energy 47(57), 23994–24003. https://doi.org/10.1016/j.ijhydene.2022.04.234. [CrossRef] [Google Scholar]
- Aakko-Saksa P.T., Vehkamäki M., Kemell M., Keskiväli L., Simell P., Reinikainen M., Tapper U., Repo T. (2020) Hydrogen release from liquid organic hydrogen carriers catalysed by platinum on rutile-anatase structured titania, Chem. Commun. 56, 11, 1657–1660. https://doi.org/10.1039/C9CC09715E. [CrossRef] [PubMed] [Google Scholar]
- Modisha P., Gqogqa P., Garidzirai R., Ouma C.N.M., Bessarabov D. (2019) Evaluation of catalyst activity for release of hydrogen from liquid organic hydrogen carriers, Int. J. Hydrogen Energy 44, 39, 21926–21935. https://doi.org/10.1016/j.ijhydene.2019.06.212. [CrossRef] [Google Scholar]
- Modisha P., Garidzirai R., Güneş H., Bozbag S.E., Rommel S., Uzunlar E., Aindow M., Erkey C., Bessarabov D. (2022) A promising catalyst for the dehydrogenation of perhydro-dibenzyltoluene: Pt/Al2O3 prepared by supercritical CO2 deposition, Catalysts 12, 5, 489. https://doi.org/10.3390/catal12050489. [CrossRef] [Google Scholar]
- Jo Y., Wan Kim T., Oh J., Kim D., Suh Y.-W. (2022) Mesoporous sulfur-decorated Pt–Al2O3 for dehydrogenation of perhydro benzyltoluenes: activity-favorable adsorption of reaction species onto electron-deficient Pt atoms, J. Catal. 413, 127–137. https://doi.org/10.1016/j.jcat.2022.06.025. [CrossRef] [Google Scholar]
- Lee S., Lee J., Kim T., Han G., Lee J., Lee K., Bae J. (2021) Pt/CeO2 catalyst synthesized by combustion method for dehydrogenation of perhydro-dibenzyltoluene as liquid organic hydrogen carrier: effect of pore size and metal dispersion, Int. J. Hydrogen Energy 46, 7, 5520–5529. https://doi.org/10.1016/j.ijhydene.2020.11.038. [CrossRef] [Google Scholar]
- Solymosi T., Geißelbrecht M., Mayer S., Auer M., Leicht P., Terlinden M., Malgaretti P., Bösmann A., Preuster P., Harting J., Thommes M., Vogel N., Wasserscheid P. (2022) Nucleation as a rate-determining step in catalytic gas generation reactions from liquid phase systems, Sci. Adv. 8, 46, eade3262. https://doi.org/10.1126/sciadv.ade3262. [CrossRef] [Google Scholar]
- Shi L., Zhou Y., Qi S., Smith K.J., Tan X., Yan J., Yi C. (2020) Pt catalysts supported on H2 and O2 plasma-treated Al2O3 for hydrogenation and dehydrogenation of the liquid organic hydrogen carrier pair dibenzyltoluene and perhydrodibenzyltoluene, ACS Catal. 10, 18, 10661–10671. https://doi.org/10.1021/acscatal.0c03091. [CrossRef] [Google Scholar]
- Ouma C.N.M., Modisha P.M., Bessarabov D. (2020) Catalytic dehydrogenation onset of liquid organic hydrogen carrier, perhydro-dibenzyltoluene: the effect of Pd and Pt subsurface configurations, Comput. Mater. Sci. 172, 109332. https://doi.org/10.1016/j.commatsci.2019.109332. [CrossRef] [Google Scholar]
- Ouma C.N.M., Modisha P., Bessarabov D. (2018) Insight into the adsorption of a liquid organic hydrogen carrier, perhydro-i-dibenzyltoluene (i = m, o, p), on Pt, Pd and PtPd planar surfaces, RSC Adv. 8, 56, 31895–31904. https://doi.org/10.1039/C8RA05800H. [CrossRef] [Google Scholar]
- Kim C.H., Lee M.-W., Jang J.S., Lee S.H., Lee K.-Y. (2021) Enhanced activity of a WOx-incorporated Pt/Al2O3 catalyst for the dehydrogenation of homocyclic LOHCs: effects of impregnation sequence on Pt–WOx interactions, Fuel 313, 8, 122654. https://doi.org/10.1016/j.fuel.2021.122654. [Google Scholar]
- Zhou J., Chung J.S., Kang S.G. (2022) Designing Pt-based subsurface alloy catalysts for the dehydrogenation of perhydro-dibenzyltoluene: a first-principles study, Appl. Surf. Sci. 579, 152142. https://doi.org/10.1016/j.apsusc.2021.152142. [CrossRef] [Google Scholar]
- Dürr S., Zilm S., Geißelbrecht M., Müller K., Preuster P., Bösmann A., Wasserscheid P. (2021) Experimental determination of the hydrogenation/dehydrogenation – equilibrium of the LOHC system H0/H18-dibenzyltoluene, Int. J. Hydrogen Energy 46(64), 32583–32594. https://doi.org/10.1016/j.ijhydene.2021.07.119. [CrossRef] [Google Scholar]
- Ali A., Khan M.A., Abbas N., Choi H. (2022) Prediction of hydrogen storage in dibenzyltoluene empowered with machine learning, J. Energy Storage 55, 105844. https://doi.org/10.1016/j.est.2022.105844. [CrossRef] [Google Scholar]
- Müller K., Aslam R., Fischer A., Stark K., Wasserscheid P., Arlt W. (2016) Experimental assessment of the degree of hydrogen loading for the dibenzyl toluene based LOHC system, Int. J. Hydrogen Energy 41, 47, 22097–22103. https://doi.org/10.1016/j.ijhydene.2016.09.196. [CrossRef] [Google Scholar]
- Ali A., Rohini A.K., Lee H.J. (2022) Dehydrogenation of perhydro-dibenzyltoluene for hydrogen production in a microchannel reactor, Int. J. Hydrogen Energy, 47(48), 20905–20914. https://doi.org/10.1016/j.ijhydene.2022.04.212. [CrossRef] [Google Scholar]
- Rüde T., Bösmann A., Preuster P., Wasserscheid P., Arlt W., Müller K. (2018) Resilience of liquid organic hydrogen carrier based energy-storage systems, Energy Technol. 6, 3, 529–539. https://doi.org/10.1002/ente.201700446. [CrossRef] [Google Scholar]
- Bulgarin A., Jorschick H., Preuster P., Bösmann A., Wasserscheid P. (2020) Purity of hydrogen released from the liquid organic hydrogen carrier compound perhydro dibenzyltoluene by catalytic dehydrogenation, Int. J. Hydrogen Energy 45, 1, 712–720. https://doi.org/10.1016/j.ijhydene.2019.10.067. [CrossRef] [Google Scholar]
- Wunsch A., Berg T., Pfeifer P. (2020) Hydrogen production from the LOHC perhydro-dibenzyl-toluene and purification using a 5 Μm PdAg-membrane in a coupled microstructured system, Materials 13, 2, 277. https://doi.org/10.3390/ma13020277. [CrossRef] [PubMed] [Google Scholar]
- Geiling J., Steinberger M., Ortner F., Seyfried R., Nuß A., Uhrig F., Lange C., Öchsner R., Wasserscheid P., März M., Preuster P. (2021) Combined dynamic operation of PEM fuel cell and continuous dehydrogenation of perhydro-dibenzyltoluene, Int. J. Hydrogen Energy, 46(72), 35662–35677. https://doi.org/10.1016/j.ijhydene.2021.08.034. [CrossRef] [Google Scholar]
- Jorschick H., Geißelbrecht M., Eßl M., Preuster P., Bösmann A., Wasserscheid P. (2020) Benzyltoluene/dibenzyltoluene-based mixtures as suitable liquid organic hydrogen carrier systems for low temperature applications, Int. J. Hydrogen Energy 45, 29, 14897–14906. https://doi.org/10.1016/j.ijhydene.2020.03.210. [CrossRef] [Google Scholar]
- Zakgeym D., Engl T., Mahayni Y., Müller K., Wolf M., Wasserscheid P. (2022) Development of an efficient Pt/SiO2 catalyst for the transfer hydrogenation from perhydro-dibenzyltoluene to acetone, Appl. Catal. A Gen. 639, 118644. https://doi.org/10.1016/j.apcata.2022.118644. [CrossRef] [Google Scholar]
- Braun K., Wolf M., De Oliveira A., Preuster P., Wasserscheid P., Thiele S., Weiß L., Wensing M. (2022) Energetics of technical integration of 2-propanol fuel cells: thermodynamic and current and future technical feasibility, Energy Technol. 10, 8, 2200343. https://doi.org/10.1002/ente.202200343. [CrossRef] [Google Scholar]
- Hauenstein P., Seeberger D., Wasserscheid P., Thiele S. (2020) High performance direct organic fuel cell using the acetone/isopropanol liquid organic hydrogen carrier system, Electrochem. Commun. 118, 106786. https://doi.org/10.1016/j.elecom.2020.106786. [CrossRef] [Google Scholar]
- Clot E., Eisenstein O., Crabtree R.H. (2007) Computational structure-activity relationships in H2 storage: How placement of N atoms affects release temperatures in organic liquid storage materials, Chem. Commun. 22, 2231–2233. https://doi.org/10.1039/B705037B. [CrossRef] [PubMed] [Google Scholar]
- Cui Y., Kwok S., Bucholtz A., Davis B., Whitney R.A., Jessop P.G. (2008) The effect of substitution on the utility of piperidines and octahydroindoles for reversible hydrogen storage, New J. Chem. 32, 6, 1027–1037. https://doi.org/10.1039/B718209K. [CrossRef] [Google Scholar]
- Sotoodeh F., Huber B.J.M., Smith K.J. (2012) The effect of the N atom on the dehydrogenation of heterocycles used for hydrogen storage, Appl. Catal. A Gen. 419–420, 67–72. https://doi.org/10.1016/j.apcata.2012.01.013. [CrossRef] [Google Scholar]
- Pez G.P., Scott A.R., Cooper A.C., Cheng H., Wilhelm F.C., Abdourazak A.H. (2008) Hydrogen storage by reversible hydrogenation of Pi-conjugated substrates, US7351395B1 April 1, 2008 (accessed 2022-11-11). https://patents.google.com/patent/US7351395B1/en [Google Scholar]
- Sotoodeh F., Smith K.J. (2013) Analysis of H2 release from organic polycyclics over Pd catalysts using DFT, J. Phys. Chem. C 117, 1, 194–204. https://doi.org/10.1021/jp307325s. [CrossRef] [Google Scholar]
- Sotoodeh F., Zhao L., Smith K.J. (2009) Kinetics of H2 Recovery from Dodecahydro-N-Ethylcarbazole over a Supported Pd Catalyst, Appl. Catal. A Gen. 362, 1, 155–162. https://doi.org/10.1016/j.apcata.2009.04.039. [CrossRef] [Google Scholar]
- Hansong Cheng. Fuel Cell Division, International Association for Hydrogen Energy (accessed 2022-11-16). https://www.iahe-fcd.org/hansong-cheng [Google Scholar]
- 武汉氢阳能源有限公司 (accessed 2022-11-16) https://www.hynertech.com/col.jsp?id=112 [Google Scholar]
- Ye X., An Y., Xu G. (2011) Kinetics of 9-ethylcarbazole hydrogenation over Raney-Ni catalyst for hydrogen storage, J. Alloys Compd. 509, 1, 152–156. https://doi.org/10.1016/j.jallcom.2010.09.012. [CrossRef] [Google Scholar]
- Wan C., An Y., Xu G., Kong W. (2012) Study of catalytic hydrogenation of N-ethylcarbazole over ruthenium catalyst, Int. J. Hydrogen Energy 37, 17, 13092–13096. https://doi.org/10.1016/j.ijhydene.2012.04.123. [CrossRef] [Google Scholar]
- Morawa Eblagon K., Tam K., Yu K.M.K., Zhao S.-L., Gong X.-Q., He H., Ye L., Wang L.-C., Ramirez-Cuesta A.J., Tsang S.C. (2010) Study of catalytic sites on ruthenium for hydrogenation of N-ethylcarbazole: implications of hydrogen storage via reversible catalytic hydrogenation, J. Phys. Chem. C 114, 21, 9720–9730. https://doi.org/10.1021/jp908640k. [CrossRef] [Google Scholar]
- Eblagon K.M., Tam K., Yu K.M.K., Tsang S.C.E. (2012) Comparative study of catalytic hydrogenation of 9-ethylcarbazole for hydrogen storage over noble metal surfaces, J. Phys. Chem. C 116, 13, 7421–7429. https://doi.org/10.1021/jp212249g. [CrossRef] [Google Scholar]
- Mehranfar A., Izadyar M., Esmaeili A.A. (2015) Hydrogen storage by N-Ethylcarbazol as a new liquid organic hydrogen carrier: a DFT study on the mechanism, Int. J. Hydrogen Energy 40, 17, 5797–5806. https://doi.org/10.1016/j.ijhydene.2015.03.011. [CrossRef] [Google Scholar]
- Liu H., Xue J., Yu P., Zhang Y., Wang J., Che D. (2023) Hydrogenation of N-ethylcarbazole with hydrogen-methane mixtures for hydrogen storage, Fuel 331, 125920. https://doi.org/10.1016/j.fuel.2022.125920. [CrossRef] [Google Scholar]
- Morawa Eblagon K., Tam K., Edman Tsang S.C. (2012) Comparison of catalytic performance of supported ruthenium and rhodium for hydrogenation of 9-ethylcarbazole for hydrogen storage applications, Energy Environ. Sci. 5, 9, 8621–8630. https://doi.org/10.1039/C2EE22066K. [CrossRef] [Google Scholar]
- Eblagon K.M., Rentsch D., Friedrichs O., Remhof A., Zuettel A., Ramirez-Cuesta A.J., Tsang S.C. (2010) Hydrogenation of 9-ethylcarbazole as a prototype of a liquid hydrogen carrier, Int. J. Hydrogen Energy 35, 20, 11609–11621. https://doi.org/10.1016/j.ijhydene.2010.03.068. [CrossRef] [Google Scholar]
- Shin B.S., Yoon C.W., Kwak S.K., Kang J.W. (2018) Thermodynamic assessment of carbazole-based organic polycyclic compounds for hydrogen storage applications via a computational approach, Int. J. Hydrogen Energy 43, 27, 12158–12167. https://doi.org/10.1016/j.ijhydene.2018.04.182. [CrossRef] [Google Scholar]
- Forberg D., Schwob T., Zaheer M., Friedrich M., Miyajima N., Kempe R. (2016) Single-Catalyst High-Weight% Hydrogen Storage in an N -Heterocycle Synthesized from Lignin Hydrogenolysis Products and Ammonia, Nature Commun. 7, 1, 1–6. https://doi.org/10.1038/ncomms13201. [CrossRef] [Google Scholar]
- Yu H., Yang X., Wu Y., Guo Y., Li S., Lin W., Li X., Zheng J. (2020) Bimetallic Ru-Ni/TiO2 catalysts for hydrogenation of N-ethylcarbazole: role of TiO2 crystal structure, J. Energy Chem. 40, 188–195. https://doi.org/10.1016/j.jechem.2019.04.009. [CrossRef] [Google Scholar]
- Qin Y., Bai X. (2022) Hydrogenation of N-ethylcarbazole over Ni-Ru alloy nanoparticles loaded on graphitized carbon prepared by carbothermal reduction, Fuel 307, 121921. https://doi.org/10.1016/j.fuel.2021.121921. [CrossRef] [Google Scholar]
- Wang Y., Bai X. (2023) Efficient catalytic hydrogen storage of N-ethylcarbazole over RuNi alloy nanoparticles loaded on SBA-15 prepared by electrostatic adsorption, Fuel 331, 125709. https://doi.org/10.1016/j.fuel.2022.125709. [CrossRef] [Google Scholar]
- Liu X., Bai X., Wu W. (2022) Ultrasound-assisted green synthesis of Ru supported on LDH-CNT composites as an efficient catalyst for N-ethylcarbazole hydrogenation, Ultrasonics Sonochem., 91, 106227. https://doi.org/10.1016/j.ultsonch.2022.106227. [CrossRef] [Google Scholar]
- Wu Y., Yu H., Guo Y., Jiang X., Qi Y., Sun B., Li H., Zheng J., Li X. (2019) A rare earth hydride supported ruthenium catalyst for the hydrogenation of N-heterocycles: boosting the activity via a new hydrogen transfer path and controlling the stereoselectivity, Chem. Sci. 10, 45, 10459–10465. https://doi.org/10.1039/C9SC04365A. [CrossRef] [PubMed] [Google Scholar]
- Wu Y., Yu H., Guo Y., Zhang Y., Jiang X., Sun B., Fu K., Chen J., Qi Y., Zheng J., Li X. (2019) Promoting hydrogen absorption of liquid organic hydrogen carriers by solid metal hydrides, J. Mater. Chem. A 7, 28, 16677–16684. https://doi.org/10.1039/C9TA05966K. [CrossRef] [Google Scholar]
- Wu Y., Guo Y., Yu H., Jiang X., Zhang Y., Qi Y., Fu K., Xie L., Li G., Zheng J., Li X. (2021) Nonstoichiometric yttrium hydride–promoted reversible hydrogen storage in a liquid organic hydrogen carrier, CCS Chem. 3, 3, 974–984. https://doi.org/10.31635/ccschem.020.202000255 [CrossRef] [Google Scholar]
- Yu H., Yang X., Jiang X., Wu Y., Chen S., Lin W., Wu Y., Xie L., Li X., Zheng J. (2021) LaNi5.5 Particles for Reversible Hydrogen Storage in N-Ethylcarbazole, Nano Energy 80, 105476. https://doi.org/10.1016/j.nanoen.2020.105476. [CrossRef] [Google Scholar]
- Yang M., Dong Y., Fei S., Ke H., Cheng H. (2014) A comparative study of catalytic dehydrogenation of perhydro-N-ethylcarbazole over noble metal catalysts, Int. J. Hydrogen Energy 39, 33, 18976–18983. https://doi.org/10.1016/j.ijhydene.2014.09.123. [CrossRef] [Google Scholar]
- Yang M., Han C., Ni G., Wu J., Cheng H. (2012) Temperature controlled three-stage catalytic dehydrogenation and cycle performance of perhydro-9-ethylcarbazole, Int. J. Hydrogen Energy 37, 17, 12839–12845. https://doi.org/10.1016/j.ijhydene.2012.05.092. [CrossRef] [Google Scholar]
- Sobota M., Nikiforidis I., Amende M., Zanón B.S., Staudt T., Höfert O., Lykhach Y., Papp C., Hieringer W., Laurin M., Assenbaum D., Wasserscheid P., Steinrück H.-P., Görling A., Libuda J. (2011) Dehydrogenation of dodecahydro-N-ethylcarbazole on Pd/Al2O3 model catalysts, Chem. A Eur. J. 17 41, 11542–11552. https://doi.org/10.1002/chem.201101311. [CrossRef] [PubMed] [Google Scholar]
- Amende M., Schernich S., Sobota M., Nikiforidis I., Hieringer W., Assenbaum D., Gleichweit C., Drescher H.-J., Papp C., Steinrück H.-P., Görling A., Wasserscheid P., Laurin M., Libuda J. (2013) Dehydrogenation mechanism of liquid organic hydrogen carriers: dodecahydro-N-ethylcarbazole on Pd(111), Chem. A Eur. J. 19, 33, 10854–10865. https://doi.org/10.1002/chem.201301323. [CrossRef] [PubMed] [Google Scholar]
- Gleichweit C., Amende M., Schernich S., Zhao W., Lorenz M.P.A., Höfert O., Brückner N., Wasserscheid P., Libuda J., Steinrück H.-P., Papp C. (2013) Dehydrogenation of dodecahydro-N-ethylcarbazole on Pt(111), ChemSusChem 6, 6, 974–977. https://doi.org/10.1002/cssc.201300263. [CrossRef] [PubMed] [Google Scholar]
- Amende M., Gleichweit C., Werner K., Schernich S., Zhao W., Lorenz M.P.A., Höfert O., Papp C., Koch M., Wasserscheid P., Laurin M., Steinrück H.-P., Libuda J. (2014) Model catalytic studies of liquid organic hydrogen carriers: dehydrogenation and decomposition mechanisms of dodecahydro-N-ethylcarbazole on Pt(111), ACS Catal. 4, 2, 657–665. https://doi.org/10.1021/cs400946x. [CrossRef] [Google Scholar]
- Amende M., Gleichweit C., Schernich S., Höfert O., Lorenz M.P.A., Zhao W., Koch M., Obesser K., Papp C., Wasserscheid P., Steinrück H.-P., Libuda J. (2014) Size and structure effects controlling the stability of the liquid organic hydrogen carrier dodecahydro-N-ethylcarbazole during dehydrogenation over Pt model catalysts, J. Phys. Chem. Lett. 5, 8, 1498–1504. https://doi.org/10.1021/jz500157r. [CrossRef] [Google Scholar]
- Kustov L.M., Tarasov A.L., Kirichenko O.A. (2017) Microwave-activated dehydrogenation of perhydro-N-ethylcarbazol over bimetallic Pd-M/TiO2 catalysts as the second stage of hydrogen storage in liquid substrates, Int. J. Hydrogen Energy 42, 43, 26723–26729. https://doi.org/10.1016/j.ijhydene.2017.09.009. [CrossRef] [Google Scholar]
- Jiang Z., Gong X., Wang B., Wu Z., Fang T. (2019) A experimental study on the dehydrogenation performance of dodecahydro-N-ethylcarbazole on M/TiO2 catalysts, Int. J. Hydrogen Energy 44, 5, 2951–2959. https://doi.org/10.1016/j.ijhydene.2018.11.236. [CrossRef] [Google Scholar]
- Wang B., Chang T., Jiang Z., Wei J., Zhang Y., Yang S., Fang T. (2018) Catalytic dehydrogenation study of dodecahydro-N-ethylcarbazole by noble metal supported on reduced graphene oxide, Int. J. Hydrogen Energy 43, 15, 7317–7325. https://doi.org/10.1016/j.ijhydene.2018.02.156. [CrossRef] [Google Scholar]
- Jiang Z., Guo S., Fang T. (2019) Enhancing the catalytic activity and selectivity of PdAu/SiO2 bimetallic catalysts for dodecahydro-N-ethylcarbazole dehydrogenation by controlling the particle size and dispersion, ACS Appl. Energy Mater. 2, 10, 7233–7243. https://doi.org/10.1021/acsaem.9b01202. [CrossRef] [Google Scholar]
- Qiao X., She T., Zhang H., Wen X., Niu L., Ricardez-Sandoval L., Li J., Bai G. (2019) One-pot synthesis of porous silica-supported ultrafine Ni nanoparticles as efficient and stable catalyst for selective hydrogenation of benzophenone, Appl. Catal. B Environ. 259, 118111. https://doi.org/10.1016/j.apcatb.2019.118111. [CrossRef] [Google Scholar]
- Ding C., Zhu T., Wang F., Zhang Z., Dong Y., Yang M., Cheng G., Ke H., Cheng H. (2020) High active Pd@mil-101 catalyst for dehydrogenation of liquid organic hydrogen carrier, Int. J. Hydrogen Energy 45, 32, 16144–16152. https://doi.org/10.1016/j.ijhydene.2020.04.081. [CrossRef] [Google Scholar]
- Gong X., Jiang Z., Fang T. (2020) Enhancing selectivity and reducing cost for dehydrogenation of dodecahydro-N-ethylcarbazole by supporting platinum on titanium dioxide, Int. J. Hydrogen Energy 45, 11, 6838–6847. https://doi.org/10.1016/j.ijhydene.2019.12.203. [CrossRef] [Google Scholar]
- Wang B., Chen Y.-T., Chang T.-Y., Jiang Z., Huang Z.-Q., Wang S.-Y., Chang C.-R., Chen Y.-S., Wei J.-J., Yang S., Fang T. (2020) Facet-dependent catalytic activities of Pd/RGO: Exploring dehydrogenation mechanism of dodecahydro-N-ethylcarbazole, Appl. Catal. B Environ. 266, 118658. https://doi.org/10.1016/j.apcatb.2020.118658. [CrossRef] [Google Scholar]
- Feng Z., Chen X., Bai X. (2020) Catalytic dehydrogenation of liquid organic hydrogen carrier dodecahydro-N-ethylcarbazole over palladium catalysts supported on different supports, Environ. Sci. Pollut. Res. 27, 36172–36185. https://doi.org/10.1007/s11356-020-09698-w. [CrossRef] [PubMed] [Google Scholar]
- Wang Y., Feng Z., Bai X. (2022) Ultrafine palladium nanoparticles supported on mesoporous silica: an outstanding catalytic activity for hydrogen production from dodecahydro-N-ethylcarbazole, Fuel 315, 123236. https://doi.org/10.1016/j.fuel.2022.123236. [CrossRef] [Google Scholar]
- Wu Y., Liu X., Bai X., Wu W. (2022) Ultrasonic-assisted preparation of ultrafine Pd nanocatalysts loaded on Cl−-intercalated MgAl layered double hydroxides for the catalytic dehydrogenation of dodecahydro-N-ethylcarbazole, Ultrason. Sonochem. 88, 106097. https://doi.org/10.1016/j.ultsonch.2022.106097. [CrossRef] [Google Scholar]
- Wang B., Chang T., Gong X., Jiang Z., Yang S., Chen Y., Fang T. (2019) One-pot synthesis of Au/Pd core/shell nanoparticles supported on reduced graphene oxide with enhanced dehydrogenation performance for dodecahydro-N-ethylcarbazole, ACS Sustain. Chem. Eng. 7, 1, 1760–1768. https://doi.org/10.1021/acssuschemeng.8b05671. [CrossRef] [Google Scholar]
- Wang B., Chang T., Jiang Z., Wei J., Fang T. (2019) Component controlled synthesis of bimetallic PdCu nanoparticles supported on reduced graphene oxide for dehydrogenation of dodecahydro-N-ethylcarbazole, Appl. Catal. B Environ. 251, 261–272. https://doi.org/10.1016/j.apcatb.2019.03.071. [CrossRef] [Google Scholar]
- Feng Z., Bai X. (2022) Enhanced activity of bimetallic Pd-Ni nanoparticles on KIT-6 for production of hydrogen from dodecahydro-N-ethylcarbazole, Fuel 329, 125473. https://doi.org/10.1016/j.fuel.2022.125473. [CrossRef] [Google Scholar]
- Eblagon K.M., Tsang S.C.E. (2015) Structure-reactivity relationship in catalytic hydrogenation of heterocyclic compounds over ruthenium black; Part B: effect of carbon substitution by heteroatom, Appl. Catal. B Environ. 163, 599–610. https://doi.org/10.1016/j.apcatb.2014.08.040. [CrossRef] [Google Scholar]
- Krishnamurthy S., Panvelker S., Shah Y.T. (1981) Hydrodeoxygenation of dibenzofuran and related compounds, AIChE J. 27, 6, 994–1001. https://doi.org/10.1002/aic.690270616. [CrossRef] [Google Scholar]
- Wang L., Zhang M., Zhang M., Sha G., Liang C. (2013) Hydrodeoxygenation of dibenzofuran over mesoporous silica COK-12 supported palladium catalysts, Energy Fuels 27, 4, 2209–2217. https://doi.org/10.1021/ef302166q. [CrossRef] [Google Scholar]
- Wang L., Li C., Jin S., Li W., Liang C. (2014) Hydrodeoxygenation of dibenzofuran over SBA-15 supported Pt, Pd, and Ru catalysts, Catal. Lett. 144, 5, 809–816. https://doi.org/10.1007/s10562-014-1236-2. [CrossRef] [MathSciNet] [Google Scholar]
- Jang M., Shin B.S., Jo Y.S., Kang J.W., Kwak S.K., Yoon C.W., Jeong H. (2020) A study on hydrogen uptake and release of a eutectic mixture of biphenyl and diphenyl ether, Eur. J. Org. Chem. 42, 11–16. https://doi.org/10.1016/j.jechem.2019.05.024. [Google Scholar]
- Morton D., Cole-Hamilton D.J. (1987) Rapid thermal hydrogen production from alcohols catalysed by [Rh(2,2’-Bipyridyl)2JCI, J. Chem. Soc., Chem. Commun. 248–249 2. https://doi.org/10.1039/C39870000248. [Google Scholar]
- Nystrom R.F., Brown W.G. (1947) Reduction of organic compounds by lithium aluminum hydride. I. Aldehydes, ketones, esters, acid chlorides and acid anhydrides, J. Am. Chem. Soc. 69, 5, 1197–1199. https://doi.org/10.1021/ja01197a060. [CrossRef] [Google Scholar]
- Taniguchi T., Curran D.P. (2012) Silica gel promotes reductions of aldehydes and ketones by N-heterocyclic carbene boranes, Org. Lett. 14, 17, 4540–4543. https://doi.org/10.1021/ol302010f. [CrossRef] [PubMed] [Google Scholar]
- Sabatier P., Senderens J.-B. (1903) Décomposition catalytique de l’alcool éthylique par les métaux divisés; formation régulière d’aldéhyde, Gallica. (accessed 2022-11-21). https://gallica.bnf.fr/ark:/12148/bpt6k3091c [Google Scholar]
- Redina E.A., Vikanova K.V., Kapustin G.I., Mishin I.V., Tkachenko O.P., Kustov L.M. (2019) Selective room-temperature hydrogenation of carbonyl compounds under atmospheric pressure over platinum nanoparticles supported on ceria-zirconia mixed oxide, Eur. J. Org. Chem. 2019, 26, 4159–4170. https://doi.org/10.1002/ejoc.201900215. [CrossRef] [Google Scholar]
- Rachmady W., Vannice M.A. (2000) Acetic acid hydrogenation over supported platinum catalysts, J. Catal. 192, 2, 322–334. https://doi.org/10.1006/jcat.2000.2863. [CrossRef] [Google Scholar]
- Pan M., Flaherty D.W., Mullins C.B. (2011) Low-temperature hydrogenation of acetaldehyde to ethanol on H-precovered Au(111), J. Phys. Chem. Lett. 2, 12, 1363–1367. https://doi.org/10.1021/jz200577n. [CrossRef] [Google Scholar]
- Chen C.-C., Lin L., Ye R.-P., Huang L., Zhu L.-B., Huang Y.-Y., Qin Y.-Y., Yao Y.-G. (2021) Construction of Cu-Ce composite oxides by simultaneous ammonia evaporation method to enhance catalytic performance of Ce-Cu/SiO2 catalysts for dimethyl oxalate hydrogenation, Fuel 290, 120083. https://doi.org/10.1016/j.fuel.2020.120083. [CrossRef] [Google Scholar]
- Ni J., Leng W., Mao J., Wang J., Lin J., Jiang D., Li X. (2019) Tuning electron density of metal nickel by support defects in Ni/ZrO2 for selective hydrogenation of fatty acids to alkanes and alcohols, Appl. Catal. B Environ. 253, 170–178. https://doi.org/10.1016/j.apcatb.2019.04.043. [CrossRef] [Google Scholar]
- Kong X., Chen L. (2014) Chemoselective hydrogenation of aromatic aldehydes over SiO2 modified Co/γ-Al2O3, Appl. Catal. A Gen. 476, 34–38. https://doi.org/10.1016/j.apcata.2014.02.011. [CrossRef] [Google Scholar]
- He Z., Lin H., He P., Yuan Y. (2011) Effect of Boric Oxide Doping On The Stability And Activity of a Cu–SiO2 catalyst for vapor-phase hydrogenation of dimethyl oxalate to ethylene glycol, J. Catal. 277, 1, 54–63. https://doi.org/10.1016/j.jcat.2010.10.010. [CrossRef] [Google Scholar]
- Zheng X., Lin H., Zheng J., Duan X., Yuan Y. (2013) Lanthanum oxide-modified Cu/SiO2 as a high-performance catalyst for chemoselective hydrogenation of dimethyl oxalate to ethylene glycol, ACS Catal. 3, 12, 2738–2749. https://doi.org/10.1021/cs400574v. [CrossRef] [Google Scholar]
- Manikandan M., Venugopal A.K., Nagpure A.S., Chilukuri S., Raja T. (2016) Promotional effect of Fe on the performance of supported Cu catalyst for ambient pressure hydrogenation of furfural, RSC Adv. 6, 5, 3888–3898. https://doi.org/10.1039/C5RA24742J. [CrossRef] [Google Scholar]
- Yu X., Vest T.A., Gleason-Boure N., Karakalos S.G., Tate G.L., Burkholder M., Monnier J.R., Williams C.T. (2019) Enhanced hydrogenation of dimethyl oxalate to ethylene glycol over indium promoted Cu/SiO2, J. Catal. 380, 289–296. https://doi.org/10.1016/j.jcat.2019.10.001. [CrossRef] [Google Scholar]
- Zhu Y., Shi L. (2014) Zn promoted Cu–Al catalyst for hydrogenation of ethyl acetate to alcohol, J. Ind. Eng. Chem. 20, 4, 2341–2347. https://doi.org/10.1016/j.jiec.2013.10.010. [CrossRef] [Google Scholar]
- Huang C., Zhang H., Zhao Y., Chen S., Liu Z. (2012) Diatomite-supported Pd–M (M=Cu Co, Ni) bimetal nanocatalysts for selective hydrogenation of long-chain aliphatic esters, J. Colloid Interface Sci. 386, 1, 60–65. https://doi.org/10.1016/j.jcis.2012.07.032. [CrossRef] [Google Scholar]
- Liu Y., Ding J., Yang J., Bi J., Liu K., Chen J. (2017) Stabilization of copper catalysts for hydrogenation of dimethyl oxalate by deposition of Ag clusters on Cu nanoparticles, Catal. Commun. 98, 43–46. https://doi.org/10.1016/j.catcom.2017.05.007. [CrossRef] [Google Scholar]
- Wang Y., Duan X., Zheng J., Lin H., Yuan Y., Ariga H., Takakusagi S., Asakura K. (2012) Remarkable enhancement of Cu catalyst activity in hydrogenation of dimethyl oxalate to ethylene glycol using gold, Catal. Sci. Technol. 2, 8, 1637–1639. https://doi.org/10.1039/C2CY20154B. [CrossRef] [Google Scholar]
- Haffad D., Kameswari U., Bettahar M.M., Chambellan A., Lavalley J.C. (1997) Reduction of benzaldehyde on metal oxides, J. Catal. 172, 1, 85–92. https://doi.org/10.1006/jcat.1997.1854. [CrossRef] [Google Scholar]
- Cox J.D., Pilcher G. (1970) Thermochemistry of organic and organometallic compounds, Academic Press (accessed 2022-11-23). https://scholar.google.com/scholar_lookup?title=Thermochemistry+of+organic+and+organometallic+compounds&author=Cox%2C+J.+D.&publication_year=1970 [Google Scholar]
- Wiberg K.B., Crocker L.S., Morgan K.M. (1991) Thermochemical studies of carbonyl compounds. 5. Enthalpies of reduction of carbonyl groups, J. Am. Chem. Soc. 113, 9, 3447–3450. https://doi.org/10.1021/ja00009a033. [CrossRef] [Google Scholar]
- Church J.M., Joshi H.K. (1951) Acetaldehyde by dehydrogenation of ethyl alcohol, Ind. Eng. Chem. 43, 8, 1804–1811. https://doi.org/10.1021/ie50500a035. [CrossRef] [Google Scholar]
- Zhang P., Wang Q.-N., Yang X., Wang D., Li W.-C., Zheng Y., Chen M., Lu A.-H. (2017) A highly porous carbon support rich in graphitic-N stabilizes copper nanocatalysts for efficient ethanol dehydrogenation, ChemCatChem 9, 3, 505–510. https://doi.org/10.1002/cctc.201601373. [CrossRef] [Google Scholar]
- Wang Q.-N., Shi L., Li W., Li W.-C., Si R., Schüth F., Lu A.-H. (2018) Cu supported on thin carbon layer-coated porous SiO2 for efficient ethanol dehydrogenation, Catal. Sci. Technol. 8, 2, 472–479. https://doi.org/10.1039/C7CY02057K. [CrossRef] [Google Scholar]
- Li M.-Y., Lu W.-D., He L., Schüth F., Lu A.-H. (2019) Tailoring the surface structure of silicon carbide support for copper catalyzed ethanol dehydrogenation, ChemCatChem 11, 1, 481–487. https://doi.org/10.1002/cctc.201801742. [CrossRef] [Google Scholar]
- Hanukovich S., Dang A., Christopher P. (2019) Influence of metal oxide support acid sites on Cu-catalyzed nonoxidative dehydrogenation of ethanol to acetaldehyde, ACS Catal. 9, 4, 3537–3550. https://doi.org/10.1021/acscatal.8b05075. [CrossRef] [Google Scholar]
- Pampararo G., Garbarino G., Riani P., Villa García M., Sánchez Escribano V., Busca G. (2020) A study of ethanol dehydrogenation to acetaldehyde over supported copper catalysts: catalytic activity, deactivation and regeneration, Appl. Catal. A Gen. 602, 117710. https://doi.org/10.1016/j.apcata.2020.117710. [CrossRef] [Google Scholar]
- Hao Y., Zhao D., Liu W., Zhang M., Lou Y., Wang Z., Tang Q., Yang J. (2022) Uniformly dispersed Cu nanoparticles over mesoporous silica as a highly selective and recyclable ethanol dehydrogenation catalyst, Catalysts 12, 9, 1049. https://doi.org/10.3390/catal12091049. [CrossRef] [Google Scholar]
- Idriss H. (2004) Ethanol reactions over the surfaces of noble metal/cerium oxide catalysts, Plat. Met. Rev. 48, 3, 105–115. https://doi.org/10.1595/147106704X1603. [CrossRef] [Google Scholar]
- Pacheco H.P., de Souza E.F., Landi S.M., David M.V., Tyler Prillaman J., Davis R.J., Toniolo F.S. (2019) Ru promoted MgO and Al-modified MgO for ethanol upgrading, Top Catal. 62, 12, 894–907. https://doi.org/10.1007/s11244-019-01177-y. [CrossRef] [Google Scholar]
- Mitsudome T., Mikami Y., Funai H., Mizugaki T., Jitsukawa K., Kaneda K. (2008) Oxidant-free alcohol dehydrogenation using a reusable hydrotalcite-supported silver nanoparticle catalyst, Angew. Chem. Int. Ed. 47, 1, 138–141. https://doi.org/10.1002/anie.200703161. [CrossRef] [PubMed] [Google Scholar]
- Cornejo-Romero J., Solis-Garcia A., Vega-Diaz S.M., Fierro-Gonzalez J.C. (2017) Reverse hydrogen spillover during ethanol dehydrogenation on TiO2-supported gold catalysts, Mole. Catal. 433, 391–402. https://doi.org/10.1016/j.mcat.2017.02.041. [CrossRef] [Google Scholar]
- Chernov A.N., Astrakova T.V., Koltunov K.Y., Sobolev V.I. (2021) Ethanol dehydrogenation to acetaldehyde over Co@N-doped carbon, Catalysts 11, 11, 1411. https://doi.org/10.3390/catal11111411. [CrossRef] [Google Scholar]
- Mamontov G.V., Grabchenko M.V., Sobolev V.I., Zaikovskii V.I., Vodyankina O.V. (2016) Ethanol dehydrogenation over Ag-CeO2/SiO2 catalyst: Role of Ag-CeO2 interface, Appl. Catal. A Gen. 528, 161–167. https://doi.org/10.1016/j.apcata.2016.10.005. [CrossRef] [Google Scholar]
- Sushkevich V.L., Ivanova I.I., Taarning E. (2013) Mechanistic study of ethanol dehydrogenation over silica-supported silver, ChemCatChem 5, 8, 2367–2373. https://doi.org/10.1002/cctc.201300033. [CrossRef] [Google Scholar]
- Wang C., Garbarino G., Allard L.F., Wilson F., Busca G., Flytzani-Stephanopoulos M. (2016) Low-temperature dehydrogenation of ethanol on atomically dispersed gold supported on ZnZrOx, ACS Catal. 6, 1, 210–218. https://doi.org/10.1021/acscatal.5b01593. [CrossRef] [Google Scholar]
- Rodriguez-Gomez A., Holgado J.P., Caballero A. (2017) Cobalt carbide identified as catalytic site for the dehydrogenation of ethanol to acetaldehyde, ACS Catal. 7, 8, 5243–5247. https://doi.org/10.1021/acscatal.7b01348. [CrossRef] [Google Scholar]
- Wadsö I., Bjerrum J., Trætteberg M., Grönvall A., Zaar B., Diczfalusy E. (1958) The heats of hydrolysis of some alkyl acetates, Acta Chem. Scand. 12, 630–634. https://doi.org/10.3891/acta.chem.scand.12-0630. [CrossRef] [Google Scholar]
- Christensen C.H., Jørgensen B., Rass-Hansen J., Egeblad K., Madsen R., Klitgaard S.K., Hansen S.M., Hansen M.R., Andersen H.C., Riisager A. (2006) Formation of acetic acid by aqueous-phase oxidation of ethanol with air in the presence of a heterogeneous gold catalyst, Angew. Chem. Int. Ed. 45, 28, 4648–4651. https://doi.org/10.1002/anie.200601180. [CrossRef] [PubMed] [Google Scholar]
- Balaraman E., Khaskin E., Leitus G., Milstein D. (2013) Catalytic transformation of alcohols to carboxylic acid salts and H2 using water as the oxygen atom source, Nat. Chem. 5, 2, 122–125. https://doi.org/10.1038/nchem.1536. [CrossRef] [PubMed] [Google Scholar]
- Li J.J. (2014) Cannizzaro reaction. In name reactions, Springer International Publishing:, Cham, pp. 106–107. https://doi.org/10.1007/978-3-319-03979-4_51. [Google Scholar]
- Hattori H. (2001) Solid base catalysts: generation of basic sites and application to organic synthesis, Appl. Catal. A Gen. 222, 1, 247–259. https://doi.org/10.1016/S0926-860X(01)00839-0. [CrossRef] [Google Scholar]
- Sawama Y., Morita K., Yamada T., Nagata S., Yabe Y., Monguchi Y., Sajiki H. (2014) Rhodium-on-carbon catalyzed hydrogen scavenger- and oxidant-free dehydrogenation of alcohols in aqueous media, Green Chem. 16, 7, 3439–3443. https://doi.org/10.1039/C4GC00434E. [CrossRef] [Google Scholar]
- Sawama Y., Morita K., Asai S., Kozawa M., Tadokoro S., Nakajima J., Monguchi Y., Sajiki H. (2015) Palladium on carbon-catalyzed aqueous transformation of primary alcohols to carboxylic acids based on dehydrogenation under mildly reduced pressure, Adv. Synth. Catal. 357, 6, 1205–1210. https://doi.org/10.1002/adsc.201401123. [CrossRef] [Google Scholar]
- Bordoloi K., Kalita G.D., Das P. (2022) Acceptorless dehydrogenation of alcohols to carboxylic acids by palladium nanoparticles supported on NiO: delving into metal-support cooperation in catalysis, Dalton Trans. 51, 25, 9922–9934. https://doi.org/10.1039/D2DT01311H. [CrossRef] [PubMed] [Google Scholar]
- Yin S., Zheng Q., Chen J., Tu T. (2022) Acceptorless dehydrogenation of primary alcohols to carboxylic acids by self-supported NHC-Ru single-site catalysts, J. Catal. 408, 165–172. https://doi.org/10.1016/j.jcat.2022.02.018. [CrossRef] [Google Scholar]
- Monda F., Madsen R. (2018) Zinc oxide-catalyzed dehydrogenation of primary alcohols into carboxylic acids, Chem. A Eur. J. 24, 67, 17832–17837. https://doi.org/10.1002/chem.201804402. [CrossRef] [PubMed] [Google Scholar]
- Li B., Fang J., Xu D., Zhao H., Zhu H., Zhang F., Dong Z. (2021) Atomically Dispersed Co clusters anchored on N-doped carbon nanotubes for efficient dehydrogenation of alcohols and subsequent conversion to carboxylic acids, ChemSusChem 14, 20, 4536–4545. https://doi.org/10.1002/cssc.202101330. [CrossRef] [PubMed] [Google Scholar]
- Chen C., Wang Z.-Q., Gong Y.-Y., Wang J.-C., Yuan Y., Cheng H., Sang W., Chaemchuen S., Verpoort F. (2021) Cobalt embedded in nitrogen-doped porous carbon as a robust heterogeneous catalyst for the atom-economic alcohol dehydrogenation to carboxylic acids, Carbon 174, 284–294. https://doi.org/10.1016/j.carbon.2020.12.040. [CrossRef] [Google Scholar]
- Mittal R., Awasthi S.K. (2022) Bimetallic oxide catalyst for the dehydrogenative oxidation reaction of alcohols: practical application in the synthesis of value-added chemicals, ACS Sustain. Chem. Eng. 10, 4, 1702–1713. https://doi.org/10.1021/acssuschemeng.1c07799. [CrossRef] [Google Scholar]
- Gao D., Feng Y., Yin H., Wang A., Jiang T. (2013) Coupling reaction between ethanol dehydrogenation and maleic anhydride hydrogenation catalyzed by Cu/Al2O3, Cu/ZrO2, and Cu/ZnO catalysts, Chem. Eng. J. 233, 349–359. https://doi.org/10.1016/j.cej.2013.08.058. [CrossRef] [Google Scholar]
- Franckaerts J., Froment G.F. (1964) Kinetic study of the dehydrogenation of ethanol, Chem. Eng. Sci. 19, 10, 807–818. https://doi.org/10.1016/0009-2509(64)85092-2. [CrossRef] [Google Scholar]
- Iwasa N., Takezawa N. (1991) Reforming of ethanol – dehydrogenation to ethyl acetate and steam reforming to acetic acid over copper-based catalysts, BCSJ 64, 9, 2619–2623. https://doi.org/10.1246/bcsj.64.2619. [CrossRef] [Google Scholar]
- Wang L., Zhu W., Zheng D., Yu X., Cui J., Jia M., Zhang W., Wang Z. (2010) Direct transformation of ethanol to ethyl acetate on Cu/ZrO2 Catalyst, Reac. Kinet. Mech. Cat. 101, 2, 365–375. https://doi.org/10.1007/s11144-010-0216-9. [CrossRef] [Google Scholar]
- Mitsudome T., Mikami Y., Ebata K., Mizugaki T., Jitsukawa K., Kaneda K. (2008) Copper nanoparticles on hydrotalcite as a heterogeneous catalyst for oxidant-free dehydrogenation of alcohols, Chem. Commun. 39, 4804–4806. https://doi.org/10.1039/B809012B. [CrossRef] [Google Scholar]
- Miura H., Nakahara K., Kitajima T., Shishido T. (2017) Concerted functions of surface acid-base pairs and supported copper catalysts for dehydrogenative synthesis of esters from primary alcohols, ACS Omega 2, 9, 6167–6173. https://doi.org/10.1021/acsomega.7b01142. [CrossRef] [PubMed] [Google Scholar]
- Inui K., Kurabayashi T., Sato S., Ichikawa N. (2004) Effective formation of ethyl acetate from ethanol over Cu-Zn-Zr-Al-O catalyst, J. Mole. Catal. A Chem. 216, 1, 147–156. https://doi.org/10.1016/j.molcata.2004.02.017. [CrossRef] [Google Scholar]
- Inui K., Kurabayashi T., Sato S. (2002) Direct synthesis of ethyl acetate from ethanol carried out under pressure, J. Catal. 212, 2, 207–215. https://doi.org/10.1006/jcat.2002.3769. [CrossRef] [Google Scholar]
- Colley S.W., Tabatabaei J., Waugh K.C., Wood M.A. (2005) The detailed kinetics and mechanism of ethyl ethanoate synthesis over a Cu/Cr2O3 catalyst, J. Catal. 236, 1, 21–33. https://doi.org/10.1016/j.jcat.2005.09.012. [CrossRef] [Google Scholar]
- Carotenuto G., Tesser R., Di Serio M., Santacesaria E. (2013) Kinetic study of ethanol dehydrogenation to ethyl acetate promoted by a copper/copper-chromite based catalyst, Catal. Today 203, 202–210. https://doi.org/10.1016/j.cattod.2012.02.054. [CrossRef] [Google Scholar]
- Li R., Zhang M., Yu Y. (2012) A DFT study on the Cu (111) surface for ethyl acetate synthesis from ethanol dehydrogenation, Appl. Surf. Sci. 258, 18, 6777–6784. https://doi.org/10.1016/j.apsusc.2012.01.171. [CrossRef] [Google Scholar]
- Finger P.H., Osmari T.A., Costa M.S., Bueno J.M.C., Gallo J.M.R. (2020) The role of the interface between cu and metal oxides in the ethanol dehydrogenation, Appl. Catal. A Gen. 589, 117236. https://doi.org/10.1016/j.apcata.2019.117236. [CrossRef] [Google Scholar]
- Freitas I.C., Gallo J.M.R., Bueno J.M.C., Marques C.M.P. (2016) The effect of Ag in the Cu/ZrO2 performance for the ethanol conversion, Top. Catal. 59, 2, 357–365. https://doi.org/10.1007/s11244-015-0439-0. [CrossRef] [Google Scholar]
- Sato A.G., Volanti D.P., de Freitas I.C., Longo E., Bueno J.M.C. (2012) Site-selective ethanol conversion over supported copper catalysts, Catal. Commun. 26, 122–126. https://doi.org/10.1016/j.catcom.2012.05.008. [CrossRef] [Google Scholar]
- Sato A.G., Volanti D.P., Meira D.M., Damyanova S., Longo E., Bueno J.M.C. (2013) Effect of the ZrO2 phase on the structure and behavior of supported Cu catalysts for ethanol conversion, J. Catal. 307, 1–17. https://doi.org/10.1016/j.jcat.2013.06.022. [CrossRef] [Google Scholar]
- Moromi S.K., Siddiki S.M.A.H., Ali M.A., Kon K., Shimizu K. (2014) Acceptorless dehydrogenative coupling of primary alcohols to esters by heterogeneous Pt catalysts, Catal. Sci. Technol. 4, 10, 3631–3635. https://doi.org/10.1039/C4CY00979G. [CrossRef] [Google Scholar]
- Ouyang M., Cao S., Yang S., Li M., Flytzani-Stephanopoulos M. (2020) Atomically dispersed Pd supported on zinc oxide for selective nonoxidative ethanol dehydrogenation, Ind. Eng. Chem. Res. 59, 6, 2648–2656. https://doi.org/10.1021/acs.iecr.9b05202. [CrossRef] [Google Scholar]
- McCullough L.R., Childers D.J., Watson R.A., Kilos B.A., Barton D.G., Weitz E., Kung H.H., Notestein J.M. (2017) Acceptorless dehydrogenative coupling of neat alcohols using group VI sulfide catalysts, ACS Sustain. Chem. Eng. 5, 6, 4890–4896. https://doi.org/10.1021/acssuschemeng.7b00303. [CrossRef] [Google Scholar]
- Gnanaprakasam B., Ben-David Y., Milstein D. (2010) Ruthenium pincer-catalyzed acylation of alcohols using esters with liberation of hydrogen under neutral conditions, Adv. Synth. Catal. 352, 18, 3169–3173. https://doi.org/10.1002/adsc.201000663. [CrossRef] [Google Scholar]
- Cheng J., Zhu M., Wang C., Li J., Jiang X., Wei Y., Tang W., Xue D., Xiao J. (2016) Chemoselective dehydrogenative esterification of aldehydes and alcohols with a dimeric rhodium(II) catalyst, Chem. Sci. 7, 7, 4428–4434. https://doi.org/10.1039/C6SC00145A. [CrossRef] [PubMed] [Google Scholar]
- Das U.K., Ben-David Y., Leitus G., Diskin-Posner Y., Milstein D. (2019) Dehydrogenative cross-coupling of primary alcohols to form cross-esters catalyzed by a manganese pincer complex, ACS Catal. 9, 1, 479–484. https://doi.org/10.1021/acscatal.8b04585. [CrossRef] [Google Scholar]
- Zhou Q.-Q., Zou Y.Q., Ben David Y., Milstein D. (2020) A reversible liquid to liquid organic hydrogen carrier system based on ethylene glycol and ethanol, Chem. Eur. J. chem.202002749. https://doi.org/10.1002/chem.202002749. [Google Scholar]
- Bechthold I., Bretz K., Kabasci S., Kopitzky R., Springer A. (2008) Succinic acid: a new platform chemical for biobased polymers from renewable resources, Chem. Eng. Technol. 31, 5, 647–654. https://doi.org/10.1002/ceat.200800063. [CrossRef] [Google Scholar]
- Javaid A., Bildea C.S. (2014) Design and control of an integrated 1,4-butanediol dehydrogenation and furfural hydrogenation plant, Chem. Eng. Technol. 37, 9, 1515–1524. https://doi.org/10.1002/ceat.201400210. [CrossRef] [Google Scholar]
- Zhu Y.-L., Xiang H.-W., Wu G.-S., Bai L., Li Y.-W. (2002) A novel route for synthesis of γ-butyrolactone through the coupling of hydrogenation and dehydrogenation, Chem. Commun. 3, 254–255. https://doi.org/10.1039/B109658N. [CrossRef] [Google Scholar]
- Knauth P., Sabbah R. (1990) Energetics of intra- and intermolecular bonds in ω-alkanediols: (II) thermochemical study of 1,2-ethanediol, 1,3-propanediol, 1,4-butanediol, and 1,5-pentanediol at 298.15 K, Struct Chem 1, 1, 43–46. https://doi.org/10.1007/BF00675783. [CrossRef] [Google Scholar]
- Koyama H. (October 16, 1990.) Production of lactone compound, JPH02255668A (accessed 2022-11-28) https://patents.google.com/patent/JPH02255668A/en?oq=H.+Koyama%2c+Daicel+Kagaku+Kougyou+K.+K.+Jpn.+Kokai+Tokkyo+Koho.+JP02-255668+(1990) [Google Scholar]
- Ichiki T., Mori K., Suzuki S., Ueno H., Kobayashi K. (May 11, 1993.) Process for the preparation of gamma-butyrolactone, US5210229A (accessed 2022-11-28). https://patents.google.com/patent/US5210229A/en [Google Scholar]
- Mercker H.J., Pape F.-F., Simon J., Henne A., Hesse M., Kohler U., Dostalek R., Erdbrugger C.F., Kratz D. (2000) Catalyst for dehydrogenating 1,4-butanediol to γ-butyrolactone, US6093677A. July 25, 2000. (accessed 2022-11-28). https://patents.google.com/patent/US6093677A/en?oq=]+H.J.+Mercker%2c+F.-F.+Pape%2c+J.+Simon%2c+A.+Henne%2c+M.+Hesse%2c+U.+Koehler%2c+R.+Dostalek%2c+C.F.+Erdbruegger%2c+D.+Kratz%2c+BASF+Aktiengesellschaft%2c+US+Patent+US6093677+(2000 [Google Scholar]
- Mimura H., 三村英之, Watanabe M., 渡辺真人 (1993) Method for producing γ-butyrolactone, JPH05286959A. November 2, 1993 (accessed 2022-11-28). https://patents.google.com/patent/JPH05286959A/en?oq=H.+Mimura%2c+M.+Watanabe%2c+Tosoh+K.+K.+Jpn.+Kokai+Tokkyo+Koho.+JP05-286959+(1993) [Google Scholar]
- Ichikawa N., Sato S., Takahashi R., Sodesawa T., Inui K. (2004) Dehydrogenative cyclization of 1,4-butanediol over copper-based catalyst, J. Mole. Catal. A Chem. 212, 1, 197–203. https://doi.org/10.1016/j.molcata.2003.10.028. [CrossRef] [Google Scholar]
- Bhanushali J.T., Prasad D., Patil K.N., Reddy K.S., Kainthla I., Rao K.S.R., Jadhav A.H., Nagaraja B.M. (2020) Tailoring the catalytic activity of basic mesoporous Cu/CeO2 catalyst by Al2O3 for selective lactonization and dehydrogenation of 1,4-butanediol to γ-butyrolactone, Catal. Commun. 143, 106049. https://doi.org/10.1016/j.catcom.2020.106049. [CrossRef] [Google Scholar]
- Zhang B., Zhu Y., Ding G., Zheng H., Li Y. (2012) Modification of the supported Cu/SiO2 catalyst by alkaline earth metals in the selective conversion of 1,4-butanediol to γ-butyrolactone, Appl. Catal. A Gen. 443–444, 191–201. https://doi.org/10.1016/j.apcata.2012.07.042. [CrossRef] [Google Scholar]
- Bhanushali J.T., Prasad D., Patil K.N., Babu G.V.R., Kainthla I., Rao K.S.R., Jadhav A.H., Nagaraja B.M. (2019) The selectively regulated vapour phase dehydrogenation of 1,4-butanediol to γ-butyrolactone employing a copper-based ceria catalyst, New J. Chem. 43, 30, 11968–11983. https://doi.org/10.1039/C9NJ03067K. [CrossRef] [Google Scholar]
- Patil K.N., Prasad D., Manoorkar V.K., Bhanushali J.T., Jadhav A.H., Nagaraja B.M. (2022) Selective vapour-phase dehydrocyclization of biomass-derived 1,4-butanediol to γ-butyrolactone over Cu/ZnAl2O4-CeO2 catalyst, J. Ind. Eng. Chem. 106, 142–151. https://doi.org/10.1016/j.jiec.2021.10.018. [CrossRef] [Google Scholar]
- Reddy K.H.P., Suh Y.-W., Anand N., Raju B.D., Rao K.S.R. (2017) Coupling of ortho-chloronitrobenzene hydrogenation with 1,4-butanediol dehydrogenation over CuMgO catalysts: a hydrogen free process, Catal. Commun. 95, 21–25. https://doi.org/10.1016/j.catcom.2017.02.029. [CrossRef] [Google Scholar]
- Nagaiah P., Venkat Rao M., Thirupathaiah K., Venkateshwarlu V., David Raju B., Rama Rao K.S. (2018) Selective vapour phase dehydrogenation of biomass-derived 1,4-butanediol to gamma butyrolactone over Cu/ZrO2 catalysts: influence of La2O3 promotor, Res. Chem. Intermed. 44, 10, 5817–5831. https://doi.org/10.1007/s11164-018-3457-2. [CrossRef] [Google Scholar]
- Hwang D.W., Kashinathan P., Lee J.M., Lee J.H., Lee U.H., Hwang J.S., Hwang Y.K., Chang J.S. (2011) Production of γ-butyrolactone from biomass-derived 1,4-butanediol over novel copper-silica nanocomposite, Green Chem. 13, 7, 1672–1675. https://doi.org/10.1039/C1GC15261K. [CrossRef] [Google Scholar]
- Raju M.A., Gidyonu P., Nagaiah P., Rao M.V., Raju B.D., Rao K.S.R. (2019) Mesoporous silica-supported copper catalysts for dehydrogenation of biomass-derived 1,4-butanediol to gamma butyrolactone in a continuous process at atmospheric pressure, Biomass Conv. Bioref. 9, 4, 719–726. https://doi.org/10.1007/s13399-019-00406-4. [CrossRef] [Google Scholar]
- Kim W.-H., Park I.S., Park J. (2006) Acceptor-free alcohol dehydrogenation by recyclable ruthenium catalyst, Org. Lett. 8, 12, 2543–2545. https://doi.org/10.1021/ol060750z. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Touchy A.S., Shimizu K. (2015) Acceptorless dehydrogenative lactonization of diols by Pt-loaded SnO2 catalysts, RSC Adv. 5, 37, 29072–29075. https://doi.org/10.1039/C5RA03337C. [CrossRef] [Google Scholar]
- Wada E., Tyagi A., Yamamoto A., Yoshida H. (2017) Dehydrogenative lactonization of diols with a platinum-loaded titanium oxide photocatalyst, Photochem. Photobiol. Sci. 16, 12, 1744–1748. https://doi.org/10.1039/C7PP00258K. [CrossRef] [PubMed] [Google Scholar]
- Jones D.T., Woods D.R. (1986) Acetone-butanol fermentation revisited, Microbiologic. Rev. 50, 4, 484–524. https://doi.org/10.1128/mr.50.4.484-524.1986. [CrossRef] [PubMed] [Google Scholar]
- Weber M., Pompetzki W., Bonmann R., Weber M. (2014) Acetone, Ullmann’s encyclopedia of industrial chemistry, John Wiley & Sons Ltd, pp. 1–19. https://doi.org/10.1002/14356007.a01_079.pub4. [Google Scholar]
- Snelson A., Skinner H.A. (1961) Heats of combustion: sec-propanol, 1,4-dioxan, 1,3-dioxan and tetrahydropyran, Trans. Faraday Soc. 57, 2125. https://doi.org/10.1039/tf9615702125. [CrossRef] [Google Scholar]
- Kim T.G., Yeo Y.K., Song H.K. (1992) Chemical heat pump based on dehydrogenation and hydrogenation of I-propanol and acetone, Inter. J. Energy Res. 16, 9, 897–916. https://doi.org/10.1002/er.4440160910. [CrossRef] [Google Scholar]
- Thonon C.I., J.C. Jungers (1949) La déshydrogénation des alcools secondaires en phase liquide sur le nickel, Bull. Soc. Chim. Belg. 58, 7–9, 331–349. https://doi.org/10.1002/bscb.19490580706. [CrossRef] [Google Scholar]
- Noda M., Shinoda S., Saito Y. (1988) Liquid-phase dehydrogenation of 2-propanol by suspended nickel fine-particle catalyst, BCSJ 61, 3, 961–965. https://doi.org/10.1246/bcsj.61.961. [CrossRef] [Google Scholar]
- Gastauer P., Prévost M. (1993) Dehydrogenation of isopropanol at low temperatures in the vapor phase as a reaction for a chemical heat pump, J. Chem. Eng. Jpn. 26, 5, 580–583. https://doi.org/10.1252/jcej.26.580. [CrossRef] [Google Scholar]
- Meng N., Shinoda S., Saito Y. (1997) Improvements on thermal efficiency of chemical heat pump involving the reaction couple of 2-propanol dehydrogenation and acetone hydrogenation, Int. J. Hydrogen Energy 22, 4, 361–367. https://doi.org/10.1016/S0360-3199(96)00084-5. [CrossRef] [Google Scholar]
- Xin F., Xu M., Huai X., Li X. (2013) Study on isopropanol–acetone–hydrogen chemical heat pump: liquid phase dehydrogenation of isopropanol using a reactive distillation column, Appl. Therm. Eng. 58, 1, 369–373. https://doi.org/10.1016/j.applthermaleng.2013.04.033. [CrossRef] [Google Scholar]
- Rioux R.M., Vannice M.A. (2003) Hydrogenation/dehydrogenation reactions: isopropanol dehydrogenation over copper catalysts, J. Catal. 216, 1, 362–376. https://doi.org/10.1016/S0021-9517(02)00035-0. [CrossRef] [Google Scholar]
- Rioux R.M., Vannice M.A. (2005) Dehydrogenation of isopropyl alcohol on carbon-supported Pt and Cu–Pt catalysts, J. Catal. 233, 1, 147–165. https://doi.org/10.1016/j.jcat.2005.04.020. [CrossRef] [Google Scholar]
- Kvande I., Chen D., Rønning M., Venvik H.J., Holmen A. (2005) Highly active Cu-based catalysts on carbon nanofibers for isopropanol dehydrogenation, Catal. Today 100, 3, 391–395. https://doi.org/10.1016/j.cattod.2004.10.027. [CrossRef] [Google Scholar]
- Said A.E.-A.A., Abd El-Wahab M.M.M., Goda M.N. (2016) Selective synthesis of acetone from isopropyl alcohol over active and stable CuO–NiO nanocomposites at relatively low-temperature, Egypt. J. Basic appl. Sci. 3, 4, 357–365. https://doi.org/10.1016/j.ejbas.2016.08.004. [Google Scholar]
- Morales-Anzures F., Salinas-Hernández P., Ornelas-Gutiérrez C., Tzompantzi-Morales F.J., Pérez-Hernández R. (2020) Synthesis by the sol-gel method and characterization of Pt-promoted CuO/TiO2-ZrO2 catalysts for decomposition of 2-propanol, Catal. Today 349, 228–234. https://doi.org/10.1016/j.cattod.2018.03.017. [CrossRef] [Google Scholar]
- Abdelhamid H.N., Goda M.N., Said A.E.-A.A. (2020) Selective dehydrogenation of isopropanol on carbonized metal-organic frameworks, Nano-Struct. Nano-Objects 24, 100605. https://doi.org/10.1016/j.nanoso.2020.100605. [CrossRef] [Google Scholar]
- Malineni J., Keul H., Möller M. (2015) An efficient N-heterocyclic carbene-ruthenium complex: application towards the synthesis of polyesters and polyamides, Macromol. Rapid Commun. 36, 6, 547–552. https://doi.org/10.1002/marc.201400699. [CrossRef] [Google Scholar]
- Gnanaprakasam B., Balaraman E., Ben-David Y., Milstein D. (2011) Synthesis of peptides and pyrazines from β-amino alcohols through extrusion of H2 catalyzed by ruthenium pincer complexes: ligand-controlled selectivity, Angew. Chem. Int. Ed. 50, 51, 12240–12244. https://doi.org/10.1002/anie.201105876. [CrossRef] [PubMed] [Google Scholar]
- Gunanathan C., Ben-David Y., Milstein D. (2007) Direct synthesis of amides from alcohols and amines with liberation of H2, Science 317, 5839, 790–792. https://doi.org/10.1126/science.1145295. [CrossRef] [PubMed] [Google Scholar]
- Ghosh S.C., Hong S.H. (2010) Simple RuCl3-catalyzed amide synthesis from alcohols and amines, Eur. J. Org. Chem. 2010, 22, 4266–4270. https://doi.org/10.1002/ejoc.201000362. [CrossRef] [Google Scholar]
- Saha B., Sengupta G., Sarbajna A., Dutta I., Bera J.K. (2014) Amide synthesis from alcohols and amines catalyzed by a RuII–N-Heterocyclic Carbene (NHC)–carbonyl complex, J. Organomet. Chem. 771, 124–130. https://doi.org/10.1016/j.jorganchem.2013.12.051. [CrossRef] [Google Scholar]
- Kar S., Xie Y., Zhou Q.Q., Diskin-Posner Y., Ben-David Y., Milstein D. (2021) Near-ambient-temperature dehydrogenative synthesis of the amide bond: mechanistic insight and applications, ACS Catal. 11, 12, 7383–7393. https://doi.org/10.1021/acscatal.1c00728. [CrossRef] [Google Scholar]
- Hu P., Fogler E., Diskin-Posner Y., Iron M.A., Milstein D. (2015) A novel liquid organic hydrogen carrier system based on catalytic peptide formation and hydrogenation, Nat. Commun. 6, 1, 1–7. https://doi.org/10.1038/ncomms7859. [CrossRef] [Google Scholar]
- Kothandaraman J., Kar S., Sen R., Goeppert A., Olah G.A., Prakash G.K.S. (2017) Efficient reversible hydrogen carrier system based on amine reforming of methanol, J. Am. Chem. Soc. 139, 7, 2549–2552. https://doi.org/10.1021/jacs.6b11637. [CrossRef] [PubMed] [Google Scholar]
- Nova A., Balcells D., Schley N.D., Dobereiner G.E., Crabtree R.H., Eisenstein O. (2010) An experimental-theoretical study of the factors that affect the switch between ruthenium-catalyzed dehydrogenative amide formation versus amine alkylation, Organometallics 29, 23, 6548–6558. https://doi.org/10.1021/om101015u. [CrossRef] [Google Scholar]
- Kumar A., Espinosa-Jalapa N.A., Leitus G., Diskin-Posner Y., Avram L., Milstein D. (2017) Direct synthesis of amides by dehydrogenative coupling of amines with either alcohols or esters: manganese pincer complex as catalyst, Angew. Chem. Int. Ed. 56, 47, 14992–14996. https://doi.org/10.1002/anie.201709180. [CrossRef] [PubMed] [Google Scholar]
- Shao Z., Li Y., Liu C., Ai W., Luo S.-P., Liu Q. (2020) Reversible interconversion between methanol-diamine and diamide for hydrogen storage based on manganese catalyzed (de)hydrogenation, Nat. Commun. 11, 1, 591. https://doi.org/10.1038/s41467-020-14380-3. [CrossRef] [Google Scholar]
- Xie Y., Hu P., Ben-David Y., Milstein D. (2019) A reversible liquid organic hydrogen carrier system based on methanol-ethylenediamine and ethylene urea, Angew. Chem. Int. Ed. 58, 15, 5105–5109. https://doi.org/10.1002/anie.201901695. [CrossRef] [PubMed] [Google Scholar]
- Bryant R.G. (2014) Polyimides, Ullmann’s encyclopedia of industrial chemistry, John Wiley & Sons Ltd, pp. 1–27. https://doi.org/10.1002/14356007.a21_253.pub2. [Google Scholar]
- Espinosa-Jalapa N.A., Kumar A., Leitus G., Diskin-Posner Y., Milstein D. (2017) Synthesis of cyclic imides by acceptorless dehydrogenative coupling of diols and amines catalyzed by a manganese pincer complex, J. Am. Chem. Soc. 139, 34, 11722–11725. https://doi.org/10.1021/jacs.7b08341. [CrossRef] [PubMed] [Google Scholar]
- Kumar A., Janes T., Espinosa-Jalapa N.A., Milstein D. (2018) Selective hydrogenation of cyclic imides to diols and amines and its application in the development of a liquid organic hydrogen carrier, J. Am. Chem. Soc. 140, 24, 7453–7457. https://doi.org/10.1021/jacs.8b04581. [CrossRef] [PubMed] [Google Scholar]
- Grellier M., Sabo-Etienne S. (2014) New perspectives in hydrogen storage based on RCH2NH2/RCN couples, Dalton Transac. 43, 17, 6283–6286. https://doi.org/10.1039/C3DT53583E. [CrossRef] [PubMed] [Google Scholar]
- Naeemi E., O’Connor D.G. (August 19, 2010.) Release and recovery from aliphatic primary amines or di-amines, US20100210878A1 (accessed 2021-03-16). https://patents.google.com/patent/US20100210878A1/en [Google Scholar]
- Naeemi E., O’Connor D.G., Naeemi M. (May 15, 2014.) Hydrogen storage system by catalytic dehydrogenation of amines, US20140134100A1 (accessed 2021-02-24). https://patents.google.com/patent/US20140134100A1/en [Google Scholar]
- Advantages | Asemblon Inc., Michael D Ramage (accessed 2022-11-18). https://www.asemblon.com/advantages. [Google Scholar]
- Sabatier P., Senderens J.-B. (1905) Application aux nitriles de la méthode d’hydrogénation directe par catalyse: synthèse d’amines primaires, secondaires et tertiaires, Comptes rendus hebdomadaires des séances de l’Académie des sciences, January 1, 1905, 482–486. [Google Scholar]
- Paal C., Gerum J. (1909) Über Katalytische Wirkungen Kolloidaler Metalle Der Platingruppe. VI, Reduktionskatalysen Mit Kolloidalem Palladium. Berichte der deutschen chemischen Gesellschaft 42, 2, 1553–1560. https://doi.org/10.1002/cber.19090420222. [CrossRef] [Google Scholar]
- Braun J.V., Blessing G., Katalytische Zobel F., Gegenwart Hydrierungen Unter Druck Bei, von Nickelsalzen, VI.: Nitrile. (1923) Berichte der deutschen chemischen Gesellschaft (A and B Series) 56, 8, 1988–2001. https://doi.org/10.1002/cber.19230560845. [CrossRef] [Google Scholar]
- Carothers W.H., Jones G.A. (1925) THE preparation of some primary amines by the catalytic reduction of nitrileS, J. Am. Chem. Soc. 47, 12, 3051–3057. https://doi.org/10.1021/ja01689a034. [CrossRef] [Google Scholar]
- Aller B.V. (1957) Cobalt hydrogenation catalysts. i. the preparation of the catalyst, J. Appl. Chem. 7, 3, 130–134. https://doi.org/10.1002/jctb.5010070307. [CrossRef] [Google Scholar]
- Adkins H. (1937) 1892-1949 reactions of hydrogen with organic compounds over copper-chromium oxide and nickel catalysts, University of Wisconsin Press (accessed 2022-11-18). https://scholar.google.com/scholar_lookup?title=Reactions+of+hydrogen+with+organic+compounds+over+copper-chromium+oxide+and+nickel+catalysts&author=Adkins%2C+Homer&publication_year=1937 [Google Scholar]
- Huang Y., Sachtler W.M.H. (1999) On the mechanism of catalytic hydrogenation of nitriles to amines over supported metal catalysts, Appl. Catal. A Gen. 182, 2, 365–378. https://doi.org/10.1016/S0926-860X(99)00035-6. [CrossRef] [Google Scholar]
- Barnett C. (1969) Hydrogenation of aliphatic nitriles over transition metal borides, Product R&D 8, 2, 145–149. https://doi.org/10.1021/i360030a009. [CrossRef] [Google Scholar]
- López-De Jesús Y.M., Johnson C.E., Monnier J.R., Williams C.T. (2010) Selective hydrogenation of benzonitrile by alumina-supported Ir–Pd catalysts, Top Catal. 53, 15, 1132–1137. https://doi.org/10.1007/s11244-010-9546-0. [CrossRef] [Google Scholar]
- Ryabchuk P., Agostini G., Pohl M.-M., Lund H., Agapova A., Junge H., Junge K., Beller M. (2018) Intermetallic nickel silicide nanocatalyst – a non-noble metal-based general hydrogenation catalyst, Sci. Adv. 4, 6, eaat0761. https://doi.org/10.1126/sciadv.aat0761. [CrossRef] [Google Scholar]
- Murugesan K., Senthamarai T., Sohail M., Alshammari S. A., Pohl, M.-M., Beller, V. Jagadeesh, R. (2018) Cobalt-based nanoparticles prepared from MOF–carbon templates as efficient hydrogenation catalysts, Chem. Sci. 9, 45, 8553–8560. https://doi.org/10.1039/C8SC02807A. [CrossRef] [PubMed] [Google Scholar]
- Segobia D.J., Trasarti A.F., Apesteguía C.R. (2015) Chemoselective hydrogenation of unsaturated nitriles to unsaturated primary amines: conversion of cinnamonitrile on metal-supported catalysts, Appl. Catal. A Gen. 494, 41–47. https://doi.org/10.1016/j.apcata.2015.01.028. [CrossRef] [Google Scholar]
- Chandrashekhar V.G., Senthamarai T., Kadam R.G., Malina O., Kašlík J., Zbořil R., Gawande M.B., Jagadeesh R.V., Beller M. (2022) Silica-supported Fe/Fe–O nanoparticles for the catalytic hydrogenation of nitriles to amines in the presence of aluminium additives, Nat. Catal. 5, 1, 20–29. https://doi.org/10.1038/s41929-021-00722-x. [Google Scholar]
- Segobia D.J., Trasarti A.F., Apesteguía C.R. (2012) Hydrogenation of nitriles to primary amines on metal-supported catalysts: highly selective conversion of butyronitrile to n-butylamine, Appl. Catal. A Gen. 445–446, 69–75. https://doi.org/10.1016/j.apcata.2012.08.006. [CrossRef] [Google Scholar]
- Liu C., Wang T. (2014) Isophthalonitrile (IPN) hydrogenation over K modified Ni–Co supported catalysts: catalyst characterization and performance evaluation, RSC Adv. 4, 109, 63725–63733. https://doi.org/10.1039/C4RA09607J. [CrossRef] [Google Scholar]
- Lévay K., Hegedűs L. (2019) Recent achievements in the hydrogenation of nitriles catalyzed by transitional metals, Curr. Org. Chem. 23, 18, 1881–1900. https://doi.org/10.2174/1385272823666191007160341. [CrossRef] [Google Scholar]
- Lévay K., Hegedűs L. (2018) Selective heterogeneous catalytic hydrogenation of nitriles to primary amines, Period. Polytech. Chem. Eng 62, 4, 476–488. https://doi.org/10.3311/PPch.12787. [Google Scholar]
- Kamiguchi S., Nakamura A., Suzuki A., Kodomari M., Nomura M., Iwasawa Y., Chihara T. (2005) Catalytic dehydrogenation of aliphatic amines to nitriles, imines, or vinylamines and dealkylation of tertiary aliphatic amines over halide cluster catalysts of group 5 and 6 transition metals, J. Catal. 230, 1, 204–213. https://doi.org/10.1016/j.jcat.2004.11.034. [CrossRef] [Google Scholar]
- Tseng K.-N.T., Rizzi A.M., Szymczak N.K. (2013) Oxidant-free conversion of primary amines to nitriles, J. Am. Chem. Soc. 135, 44, 16352–16355. https://doi.org/10.1021/ja409223a. [CrossRef] [PubMed] [Google Scholar]
- Wang Z., Belli J., Jensen M. C., (2011) Homogeneous dehydrogenation of liquid organic hydrogen carriers catalyzed by an iridium PCP complex, Faraday Discuss. 151, 297–305. https://doi.org/10.1039/C1FD00002K. [CrossRef] [PubMed] [Google Scholar]
- Dutta I., Yadav S., Sarbajna A., De S., Hölscher M., Leitner W., Bera J.K. (2018) Double dehydrogenation of primary amines to nitriles by a ruthenium complex featuring pyrazole functionality, J. Am. Chem. Soc. 140, 28, 8662–8666. https://doi.org/10.1021/jacs.8b05009. [CrossRef] [PubMed] [Google Scholar]
- Hale L.V.A., Malakar T., Tseng K.-N.T., Zimmerman P.M., Paul A., Szymczak N.K. (2016) The mechanism of acceptorless amine double dehydrogenation by N, N, N-amide ruthenium(II) hydrides: a combined experimental and computational study, ACS Catal. 6, 8, 4799–4813. https://doi.org/10.1021/acscatal.6b01465. [CrossRef] [Google Scholar]
- Kannan M., Muthaiah S. (2019) Extending the chemistry of hexamethylenetetramine in ruthenium-catalyzed amine oxidation, Organometallics 38, 19, 3560–3567. https://doi.org/10.1021/acs.organomet.9b00399. [CrossRef] [Google Scholar]
- Kannan M., Barteja P., Devi P., Muthaiah S. (2020) Acceptorless dehydrogenation of amines and alcohols using simple ruthenium chloride, J. Catal. 386, 1–11. https://doi.org/10.1016/j.jcat.2020.03.025. [CrossRef] [Google Scholar]
- Kannan M., Muthaiah S. (2020) Ruthenium(II)-complex-catalyzed acceptorless double dehydrogenation of primary amines to nitriles, Synlett 31, 11, 1073–1076. https://doi.org/10.1055/s-0040-1708016. [CrossRef] [Google Scholar]
- Nie X., Zheng Y., Ji L., Fu H., Chen H., Li R. (2020) Acceptorless dehydrogenation of amines to nitriles catalyzed by N-heterocyclic carbene-nitrogen-phosphine chelated bimetallic ruthenium (II) complex, J. Catal. 391, 378–385. https://doi.org/10.1016/j.jcat.2020.09.005. [CrossRef] [Google Scholar]
- Lu G.-P., Li X., Zhong L., Li S., Chen F. (2019) Ru@UiO-66(Ce) catalyzed acceptorless dehydrogenation of primary amines to nitriles: the roles of Lewis acid-base pairs in the reaction, Green Chem. 21, 19, 5386–5393. https://doi.org/10.1039/C9GC02181G. [CrossRef] [Google Scholar]
- Feldhues U., Schäfer H.J. (1982) Oxidation of primary aliphatic amines to nitriles at the nickel hydroxide electrode, Synthesis 1982, 2, 145–146. https://doi.org/10.1055/s-1982-29721. [CrossRef] [Google Scholar]
- Huang Y., Chong X., Liu C., Liang Y., Zhang B. (2018) Boosting hydrogen production by anodic oxidation of primary amines over a NiSe nanorod electrode, Angew. Chem. Int. Ed. 57, 40, 13163–13166. https://doi.org/10.1002/anie.201807717. [CrossRef] [PubMed] [Google Scholar]
- Mondal I., Hausmann J.N., Vijaykumar G., Mebs S., Dau H., Driess M., Menezes P.W. (2022) Nanostructured intermetallic nickel silicide (pre)catalyst for anodic oxygen evolution reaction and selective dehydrogenation of primary amines, Adv. Energy Mater. 12, 25, 2200269. https://doi.org/10.1002/aenm.202200269. [CrossRef] [Google Scholar]
- Qian W., Yoda Y., Hirai Y., Ishihara A., Kabe T. (1999) Hydrodesulfurization of dibenzothiophene and hydrogenation of phenanthrene on alumina-supported Pt and Pd catalysts, Appl. Catal. A Gen. 184, 1, 81–88. https://doi.org/10.1016/S0926-860X(99)00083-6. [CrossRef] [Google Scholar]
- Navarro R.M., Pawelec B., Trejo J.M., Mariscal R., Fierro J.L.G. (2000) Hydrogenation of aromatics on sulfur-resistant PtPd bimetallic catalysts, J. Catal. 189, 1, 184–194. https://doi.org/10.1006/jcat.1999.2693. [CrossRef] [MathSciNet] [Google Scholar]
- Ratner B.D., Naeemi E. (March 6, 2007.) Method for hydrogen storage and delivery, US7186396B2 (accessed 2022-11-30). https://patents.google.com/patent/US7186396/en [Google Scholar]
- Zhao H.Y., Oyama S.T., Naeemi E.D. (2010) Hydrogen storage using heterocyclic compounds: the hydrogenation of 2-methylthiophene, Catal. Today 149, 1, 172–184. https://doi.org/10.1016/j.cattod.2009.02.039. [CrossRef] [Google Scholar]
- Luo J., Rauch M., Avram L., Ben-David Y., Milstein D. (2020) Catalytic hydrogenation of thioesters, thiocarbamates, and thioamides, J. Am. Chem. Soc. 142, 52, 21628–21633. https://doi.org/10.1021/jacs.0c10884. [CrossRef] [PubMed] [Google Scholar]
- Luo J., Rauch M., Avram L., Diskin-Posner Y., Shmul G., Ben-David Y., Milstein D. (2020) Formation of thioesters by dehydrogenative coupling of thiols and alcohols with H2 evolution, Nat. Catal. 3, 11, 887–892. [CrossRef] [Google Scholar]
- Rauch M., Luo J., Avram L., Ben-David Y., Milstein D. (2021) Mechanistic investigations of ruthenium catalyzed dehydrogenative thioester synthesis and thioester hydrogenation, ACS Catal. 11, 5, 2795–2807. https://doi.org/10.1021/acscatal.1c00418. [CrossRef] [Google Scholar]
- Luo W., Zakharov L.N., Liu S.-Y. (2011) 1,2-BN cyclohexane: synthesis, structure, dynamics, and reactivity, J. Am. Chem. Soc. 133, 33, 13006–13009. https://doi.org/10.1021/ja206497x. [CrossRef] [PubMed] [Google Scholar]
- Müller K., Stark K., Müller B., Arlt W. (2012) Amine borane based hydrogen carriers: an evaluation, Energy Fuels 26, 6, 3691–3696. https://doi.org/10.1021/ef300516m. [CrossRef] [Google Scholar]
- Campbell P.G., Zakharov L.N., Grant D.J., Dixon D.A., Liu S.-Y. (2010) Hydrogen storage by boron–nitrogen heterocycles: a simple route for spent fuel regeneration, J. Am. Chem. Soc. 132, 10, 3289–3291. https://doi.org/10.1021/ja9106622. [CrossRef] [PubMed] [Google Scholar]
- Liu S.-Y. (2015) Hydrogen Storage by Novel CBN Heterocycle Materials, DE-FG36-08GO18143. Univ. of Oregon, Eugene, OR (United States). https://doi.org/10.2172/1221989 [CrossRef] [Google Scholar]
- Dai Y., Zhang X., Liu Y., Yu H., Su W., Zhou J., Ye Q., Huang Z. (2022) 1,6;2,3-Bis-BN cyclohexane: synthesis, structure, and hydrogen release, J. Am. Chem. Soc. 144, 19, 8434–8438. https://doi.org/10.1021/jacs.1c13581. [CrossRef] [PubMed] [Google Scholar]
- Technologies. HSL TECH (accessed 2023-08-31). https://www.hsl.tech/technologies. [Google Scholar]
- Finholt A.E., Jr Bond A.C., Wilzbach K.E., Schlesinger H.I. (1947) The preparation and some properties of hydrides of elements of the fourth group of the periodic system and of their organic derivatives, J. Am. Chem. Soc. 69, 11, 2692–2696. https://doi.org/10.1021/ja01203a041. [CrossRef] [Google Scholar]
- Aoyagi K., Ohmori Y., Inomata K., Matsumoto K., Shimada S., Sato K., Nakajima Y. (2019) Synthesis of hydrosilanes via lewis-base-catalysed reduction of alkoxy silanes with NaBH4, Chem. Commun. 55, 42, 5859–5862. https://doi.org/10.1039/C9CC01961H. [CrossRef] [PubMed] [Google Scholar]
- Durin G., Berthet J.-C., Nicolas E., Thuéry P., Cantat T. (2022) The role of (TBuPOCOP)Ir(I) and Iridium(III) pincer complexes in the catalytic hydrogenolysis of silyl triflates into hydrosilanes, Organometallics 41, 14, 1786–1796. https://doi.org/10.1021/acs.organomet.1c00576. [CrossRef] [Google Scholar]
- Mitsudome T., Arita S., Mori H., Mizugaki T., Jitsukawa K., Kaneda K. (2008) Supported silver-nanoparticle-catalyzed highly efficient aqueous oxidation of phenylsilanes to silanols, Angew. Chem. Int. Ed. 47, 41, 7938–7940. https://doi.org/10.1002/anie.200802761. [CrossRef] [PubMed] [Google Scholar]
- Han W.-S., Kim T.-J., Kim S.-K., Kim Y., Kim Y., Nam S.-W., Kang S.O. (2011) Silane-based hydrogen storage materials for fuel cell application: hydrogen release via methanolysis and regeneration by hydride reduction from organosilanes, Int. J. Hydrogen Energy 36, 19, 12305–12312. https://doi.org/10.1016/j.ijhydene.2011.06.118. [CrossRef] [Google Scholar]
- Brunel J.M. (2010) New efficient hydrogen process production from organosilane hydrogen carriers derivatives, Int. J. Hydrogen Energy 35, 8, 3401–3405. https://doi.org/10.1016/j.ijhydene.2010.01.116. [CrossRef] [Google Scholar]
- Mukherjee D., Thompson R.R., Ellern A., Sadow A.D. (2011) Coordinatively saturated tris(oxazolinyl)borato zinc hydride-catalyzed cross dehydrocoupling of silanes and alcohols, ACS Catal. 1, 7, 698–702. https://doi.org/10.1021/cs2001016. [CrossRef] [Google Scholar]
- Ventura-Espinosa D., Carretero-Cerdán A., Baya M., García H., Mata J.A. (2017) Catalytic dehydrogenative coupling of hydrosilanes with alcohols for the production of hydrogen on-demand: application of a silane/alcohol pair as a liquid organic hydrogen carrier, Chem. A Eur. J. 23, 45, 10815–10821. https://doi.org/10.1002/chem.201700243. [CrossRef] [PubMed] [Google Scholar]
- Ventura-Espinosa D., Sabater S., Carretero-Cerdán A., Baya M., Mata J.A. (2018) High production of hydrogen on demand from silanes catalyzed by iridium complexes as a versatile hydrogen storage system, ACS Catal. 8, 3, 2558–2566. https://doi.org/10.1021/acscatal.7b04479. [CrossRef] [Google Scholar]
- Porcar R., Mollar-Cuni A., Ventura-Espinosa D., Luis V., S. E., García-Verdugo, A. Mata, J. A Simple, (2022) Safe and robust system for hydrogenation “without high-pressure gases” under batch and flow conditions using a liquid organic hydrogen carrier, Vol. 24, Green Chemistry, pp. 2036–2043. https://doi.org/10.1039/D1GC03850H. [Google Scholar]
- Dai Y., Xing P., Cui X., Li Z., Zhang X. (2019) Coexistence of Cu(II) and Cu(I) in Cu ion-doped Zeolitic Imidazolate Frameworks (ZIF-8) for the dehydrogenative coupling of silanes with alcohols, Dalton Trans. 48, 44, 16562–16568. https://doi.org/10.1039/C9DT03181B. [CrossRef] [PubMed] [Google Scholar]
- Deyko G.S., Glukhov L.M., Kustov L.M. (2020) Hydrogen storage in organosilicon ionic liquids, Int. J. Hydrogen Energy 45, 58, 33807–33817. https://doi.org/10.1016/j.ijhydene.2020.09.107. [CrossRef] [Google Scholar]
- Schwarz D.E., Cameron T.M., Jeffrey Hay P., Scott B.L., Tumas W., Thorn D.L. (2005) Hydrogen evolution from organic “hydrides”, Chem. Commun. 47, 5919–5921. https://doi.org/10.1039/B511884K. [CrossRef] [Google Scholar]
- Proceedings of the 2000 U.S. DOE Hydrogen Program Review. 995. [Google Scholar]
- Anastas P., Eghbali N. (2010) Green chemistry: principles and practice, Chem. Soc. Rev. 39, 1, 301–312. https://doi.org/10.1039/B918763B. [CrossRef] [PubMed] [Google Scholar]
- Brigljević B., Lee B., Dickson R., Kang S., Liu J.J., Lim H. (2020) Concept for temperature-cascade hydrogen release from organic liquid carriers coupled with SOFC power generation, Cell Rep. Phys. Sci. 1, 3, 100032. https://doi.org/10.1016/j.xcrp.2020.100032. [CrossRef] [Google Scholar]
All Tables
Comparison of different LOHC over various secondary criteria. Green signifies that the LOHC complies with the criterion, red that it does not.
All Figures
|
Figure 1 Direct and indirect CO2 emission factors for different types of power generation systems obtained by life-cycle analysis and adapted from [14, 15]. |
| In the text | |
 |
Figure 2 French electricity mix from 1990 to 2021, adapted from [17]. |
| In the text | |
|
Figure 3 Energy production in France in January 2022 (left) and July 2022 (right), adapted from [20]. |
| In the text | |
|
Figure 4 Example of an energy system to produce heat through a heater from coal. |
| In the text | |
|
Figure 5 Example of an energy system to produce heat from a heater from solar radiation. |
| In the text | |
|
Figure 6 Transport range and storage time of energy vectors. Electricity and radiation cannot be directly stored and thus are not included in the diagram. |
| In the text | |
|
Figure 7 Number of policies around the world supporting hydrogen deployment, International Energy Agency, 2018 [38]. |
| In the text | |
 |
Figure 8 Feedstock distribution for the production of H2, adapted from [42, 43]. |
| In the text | |
|
Figure 10 Reviewed H2 storage technologies. |
| In the text | |
 |
Figure 11 Key steps of the circular hydrogen carriers value chain [172]. |
| In the text | |
 |
Figure 12 Key steps of the liquid organic hydrogen carrier value chain [172]. |
| In the text | |
|
Figure 13 Dehydrogenation enthalpies calculated by the PM3 semi-empirical method as a function of the number of fused rings [219]. |
| In the text | |
|
Figure 14 Clar structures for 4 fused rings: anthracene-type (left), phenanthrene-type (middle), and pyrene-type (type). |
| In the text | |
|
Figure 15 Influence of the size of the ring, the presence of N atoms, and their number on the enthalpy of dehydrogenation (kJ/molH2) [325]. |
| In the text | |
|
Figure 16 Examples of structures studied by Cooper From left to right: carbazole, indolo[3,2,1-jk]carbazole, indole, pyrroloindole, bis-indolylmethane and pyrrocoline. R= H, alkyle [219]. |
| In the text | |
|
Figure 17 Isomer structures of the 12H-NEC. The most stable is the isomer A, while the least stable is the isomer B. The structures A to D are symmetric while the structures E and F are asymmetric. |
| In the text | |
|
Figure 18 Examples of structures studied by Cooper From left to right: 4,7-phenantroline, 4,4-dipyridil, 2,2’-bipyrimidine, quinazoline, 4,2’:5’,4’’-terpyridine, 4,2’:6’,4’’-terpyridine and 1,5-naphthyridine [219]. |
| In the text | |
|
Figure 19 Comparison of the inclusion of heteroatoms on the dehydrogenation enthalpy (kJ/molH2) [219]. |
| In the text | |
|
Figure 20 Simplified mechanism of the hydrogenation followed by the hydrodeoxygenation of dibenzofuran. |
| In the text | |
|
Figure 21 Classic ligands used in homogeneous catalytic systems for the acceptorless dehydrogenation reaction. R1, R2, R3, R4 = H, alkyls, aromatics. |
| In the text | |
|
Figure 22 O and N mixed moieties obtained by the coupling of alcohols and amines |
| In the text | |
|
Figure 23 Comparison of each chemical-based H2 storage system based on their gravimetric and volumetric H2 densities. The dotted lines represent the gravimetric and volumetric densities required by the US DOE for H2 storage. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.